qPCR of poked and injected D. innubila with 16Cq DiNV fluid at different dilutions
We want to know how much more virus the flies are receiving when getting the injection compared to the needle poke. I had poked 10 and injected 10 males and immediately froze them for this. PCR information on these samples can be found here, and the day they were infected is here. I also wanted to test different dilutions, because we had some issues before where samples that should (and by all other measures were) be virus negative, but still had some viral product amplification. Potentially that was just spurious amplification at higher cycles, and that diluting the DNA will have those not show up.
Before the day of qPCR I Qubited every sample, then diluted them to 1ng/ul, 0.1ng/ul, and 0.01ng/ul. I also did a 1:10 and 1:100 dilution of the stock (non-diluted) DNA to test as well, to see if the Qubit and exact dilution is necessary.
| tube number | treatment | Qubit | dilution to 1ng/ul |
|---|---|---|---|
| 6 | 16Cq DiNV needle poke | 13.3 | 39.9 |
| 7 | 16Cq DiNV needle poke | 28.4 | 85.2 |
| 8 | 16Cq DiNV needle poke | 31.7 | 95.1 |
| 9 | 16Cq DiNV needle poke | 17.2 | 51.6 |
| 10 | 16Cq DiNV needle poke | 19.8 | 59.4 |
| 11 | 16Cq DiNV needle poke | 18.4 | 55.2 |
| 12 | 16Cq DiNV needle poke | 13.9 | 41.7 |
| 13 | 16Cq DiNV needle poke | 30.5 | 91.5 |
| 14 | 16Cq DiNV needle poke | 25.3 | 75.9 |
| 15 | 16Cq DiNV needle poke | 27.2 | 81.6 |
| 16 | 16Cq DiNV injection | 27.4 | 82.2 |
| 17 | 16Cq DiNV injection | 12.1 | 36.3 |
| 18 | 16Cq DiNV injection | 16.7 | 50.1 |
| 19 | 16Cq DiNV injection | 16.3 | 48.9 |
| 20 | 16Cq DiNV injection | 16.7 | 50.1 |
| 21 | 16Cq DiNV injection | 21.7 | 65.1 |
| 22 | 16Cq DiNV injection | 22.1 | 66.3 |
| 23 | 16Cq DiNV injection | 21.3 | 63.9 |
| 24 | 16Cq DiNV injection | 32 | 96 |
| 25 | 16Cq DiNV injection | 17.4 | 52.2 |
I used 3ul of the stock for each sample, then the final column of the table above in molecular grade water. For the 0.1ng, I did 3ul of the 1ng in 27ul of molecular grade water. For the 0.01ng, I did 3ul of the 0.1ng in 27ul of molecular grade water. For the 1:10, I did 3ul of the stock in 27ul molecular grade water. And for the 1:100, I did 3ul of the 1:10 in 27ul of molecular grade water.
All of these were made in strip tubes and frozen at -20 until use.
Only 3 samples from each treatment were used for the qPCR, along with one control sample that was a stock innubila (DNA extracted here, #8), which was diluted to 1ng/ul for use.
Here is the qPCR layout:
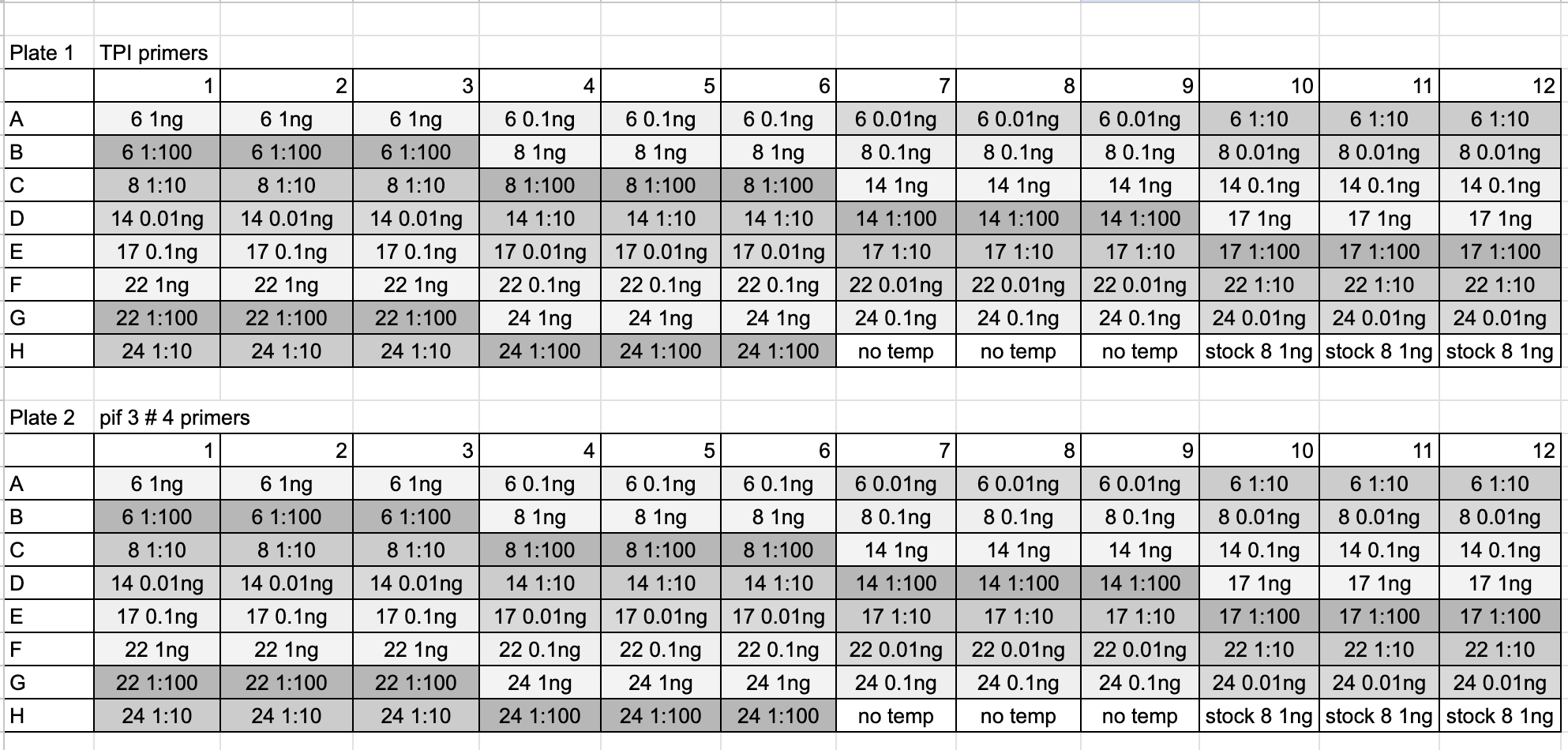
- Samples and reagents were thawed on ice, vortexed, and spun down before use
- There were 96 individual wells for each primer, 4 were added on for pipetting error
- PIF3 master mix:
- 5ul Sso supermix * 100 = 500ul
- 0.5ul PIF3 F primer * 100 = 50ul
- 0.5ul PIF3 R primer * 100 = 50ul
- 3ul nuclease free water * 100 = 300ul
- The master mix was made on ice, vortexed, and spun down
- TPI master mix:
- 5ul Sso supermix * 100 = 500ul
- 0.5ul TPI F primer * 100 = 50ul
- 0.5ul TPI R primer * 100 = 50ul
- 3ul nuclease free water * 100 = 300ul
- The master mix was made on ice, vortexed, and spun down
- I used the qPCR specific plates and seals
- 9ul of the appropriate master mix was added to the planned well (see layout above)
- 1ul of DNA was added to the planned well (see table above)
- Each well was pipette mixed with 5ul using a multichannel
- The plate was sealed and centrifuged for 6 minutes at 3,000g
- The plate was put in the qPCR machine:
- Used program CFX manager
- Selected user-defined protocol, existing protocol, and KMM folder
- Selected the p47 program
- Used the machine buttons to open and close the lid
- Started the program
- Data from the qPCR machine can be found here
- And analysis of the Cq results can be found here as a markdown