
Acropora hyacinthus Larvae Test DNA/RNA Extraction
Testing DNA/RNA Extraction and Bead or Pestle Homogenization Protocol on Acropora hyacinthus Larvae
Goal: Develop a good protocol and homogenization method for coral larvae/eggs preserved in DNA/RNA Shield. The goal is to get high quality and amount of DNA and RNA from these samples
Results: Low quality RNA, pretty degraded, but purity is ok and amount is good. DNA amount and quality pretty good. No difference in quality between homogenization methods, although yield is higher for the pestle smashed samples
Take-aways: Neither method seems good so far considering the poor RNA quality. Potentially not all the samples were preserved properly (see below), and that could have caused degradation prior to extraction. Additionally poor homogenization in both tests could have caused problems. However homogenization was worse in the pestle samples, and the quality is the same. Or incubation with proK could have caused degradation, however it was at room temp.
Samples and Protocol Notes
- Samples used are Arcopora hyacinthus larvae collected in Mo’orea on 20201112, stored in 1.5mL tubes and 1000ul of DNA/RNA Sheild, frozen at -80:
- A5_1
- A5_2
- A5_3
- A5_4
- My goal was to follow the Zymo DNA/RNA Miniprep Plus kit protocol. This is the most recent version, updated in January 2021, suggests homogenization with beads and a room temp incubation with proteinase K for 30 minutes (could be extended). Previous extractions used heated digestions with proteinase K, however Zymo has changed their product since then and the heated digestions have failed recently (probably since they changed the protocol or product. However all the buffers are named the same so it’s difficult to tell what changed).
- I also tried two different methods for homogenization: bead beating in the Tissuelyser with glass beads, and using a small disposable plastic pestle to smash the larvae by hand
Homogenization
- Thawed samples on ice bucket
- I noticed that in each sample, some of the larvae were stuck up on the side of the tube not in the shield liquid:
A5_1
A5_2
A5_3

A5_4
- I noticed that in each sample, some of the larvae were stuck up on the side of the tube not in the shield liquid:
- After thawing, poured 1/2 of a glass bead tube into each of A5_1 and A5_2
- Homogenized A5_1 and A5_2 in the Tissuelyser at 25Hz for 2 minutes
- After checking, there still seemed to be tissue left un-broken up in the samples. Additionally A5_2 had opened a little in the machine and lost some volume

- Homogenized the two tubes again for 2 minutes at 25Hz
- Homogenization seemed more complete and there were less flakes in the tubes, bubbles had to be centrifuged down in the minifuge

- Cleaned 2 plastic pestles with 10% bleach, DI water, 70% ethanol, and RNaseZap
- Used the cleaned pestles to hand-homogenize samples A5_3 and A5_4. I spent ~3-4 minutes on each tube: smashing, swirling, etc. The larvae float so it was hard to break them down. Additionally the liquid volume was a little high and some bubbled over onto my gloves. Gloves were changed between samples. After 4 minutes on each sample, I decided that was a reasonable effort even though not all of it had broken up.
A5_3

A5_4

- To each tube: added 100ul of pro K digestion buffer and 50ul of proteinase K
- Vortexed and spun down tubes. Here I noticed that the pestle homogenized tubes were less broken down than I had thought. Because they got so bubbly, I did not see un-broken up larvae floating at the top of the tubes
- Placed the tubes in the thermomixer set to 23 degrees C this is room temp, and 800rpm for 30 minutes
- After pro k incubation, the samples looked more broken down
A5_1

A5_2

A5_3

A5_4

- I wanted to try to pellet any left over debris, so I centrifuged the tubes for 1 minute at 5,000rcf (g)
- After centrifugation, there was a visible fatty layer of debris at the liquid line in each tube, as well as some pelleted debris:
A5_1

A5_2

A5_3

A5_4

- I transferred the “supernatant” to new 5mL tubes, trying to avoid the bottom and top debris. The lipid layer at the top did get partially sucked up in all samples. Volumes from each sample (note A5_2 and pestle tubes had lost volume during homogenization):
- 1: 1000ul
- 2: 700ul
- 3: 950ul
- 4: 900ul
- The previous tubes were discarded
DNA Extraction
- Warmed 10mM Tris HCl and ultrapure water in thermomixer
- Added equal volume of DNA/RNA lysis buffer to each 5mL tube
- Vortexted and spun down tubes
- Added 700ul of that liquid to a yellow spin column for each sample
- Centrifuged at 16,000 rcf for 30 seconds
- Pipetted off the flowthrough into new 5mL tubes for each sample for RNA
- Repeated last three steps three more times until all the liquid had gone through the yellow spin columns for each sample
- Added 400ul prep buffer to each spin column
- Centrifuged at 16,000 rcf for 30 seconds
- Poured off flowthrough into kit waste beaker
- Added 700ul wash buffer to each spin column
- Centrifuged at 16,000 rcf for 30 seconds
- Poured off flowthrough into kit waste beaker
- Added 400ul wash buffer to each spin column
- Centrifuged for 2 minutes at 16,000 rcf
- Transferred the spin columns to new labeled 1.5mL tubes
- Added 50ul warmed 10mM Tris HCl to each column
- Incubated at room temp for 5 minutes
- Centrifuged at 16,000 rcf for 30 seconds
- Again added 50ul warmed 10mM Tris HCl to each column
- Incubated at room temp for 5 minutes
- Centrifuged at 16,000 rcf for 30 seconds
- Saved 10ul in strip tubes for QC, froze the rest in the -20
RNA Extraction
- Added equal volume fresh 100% ethanol to each of the flow through 5mL tubes from the DNA extraction:
- 1: 2mL
- 2: 1.4mL
- 3: 1.9mL
- 4: 1.8mL
- Vortexed and spun down tubes
- Added 700ul of that liquid to a green spin column for each sample
- Centrifuged at 16,000 rcf for 30 seconds
- Poured off flowthrough into kit waste beaker
- Repeated last three steps 5 times until all of the liquid had gone through the columns
- Added 400ul wash buffer to each spin column
- Centrifuged at 16,000 rcf for 30 seconds
- Poured off flowthrough into kit waste beaker
- Created the DNase mix:
- 4 * 75ul DNA digestion buffer = 300ul
- 4 * 5ul DNase I = 20ul
- Added 80ul of the DNase mix to each tube and incubated at room temp for 15 minutes
- Added 700ul wash buffer to each spin column
- Centrifuged at 16,000 rcf for 30 seconds
- Poured off flowthrough into kit waste beaker
- Added 400ul wash buffer to each spin column
- Centrifuged for 2 minutes at 16,000 rcf
- Transferred the spin columns to new labeled 1.5mL tubes
- Added 50ul warmed ultrapure water to each column
- Incubated at room temp for 5 minutes
- Centrifuged at 16,000 rcf for 30 seconds
- Again added 50ul warmed ultrapure water to each column
- Incubated at room temp for 5 minutes
- Centrifuged at 16,000 rcf for 30 seconds
- Saved 7ul in strip tubes for QC, the rest was frozen at -80 (Silver freezer, rack D column 3)
QC
Qubit
- Broad Range dsDNA and RNA Qubit protocol
- All samples were read twice
- DNA:
| Sample | Standard 1 | Standard 2 | DNA 1 ng/ul | DNA 2 ng/ul | Average ng/ul |
|---|---|---|---|---|---|
| A5_1 | 175 | 18692 | 15.6 | 15.2 | 15.4 |
| A5_2 | - | - | 8.32 | 8.18 | 8.25 |
| A5_3 | - | - | 46.4 | 45.8 | 46.2 |
| A5_4 | - | - | 22.8 | 22.6 | 22.4 |
- RNA:
| Sample | Standard 1 | Standard 2 | RNA 1 ng/ul | RNA 2 ng/ul | Average ng/ul |
|---|---|---|---|---|---|
| A5_1 | 385 | 8326 | 89.4 | 89.2 | 89.3 |
| A5_2 | - | - | 64 | 63.2 | 63.6 |
| A5_3 | - | - | 179 | 178 | 178.5 |
| A5_4 | - | - | 94.2 | 93.8 | 93 |
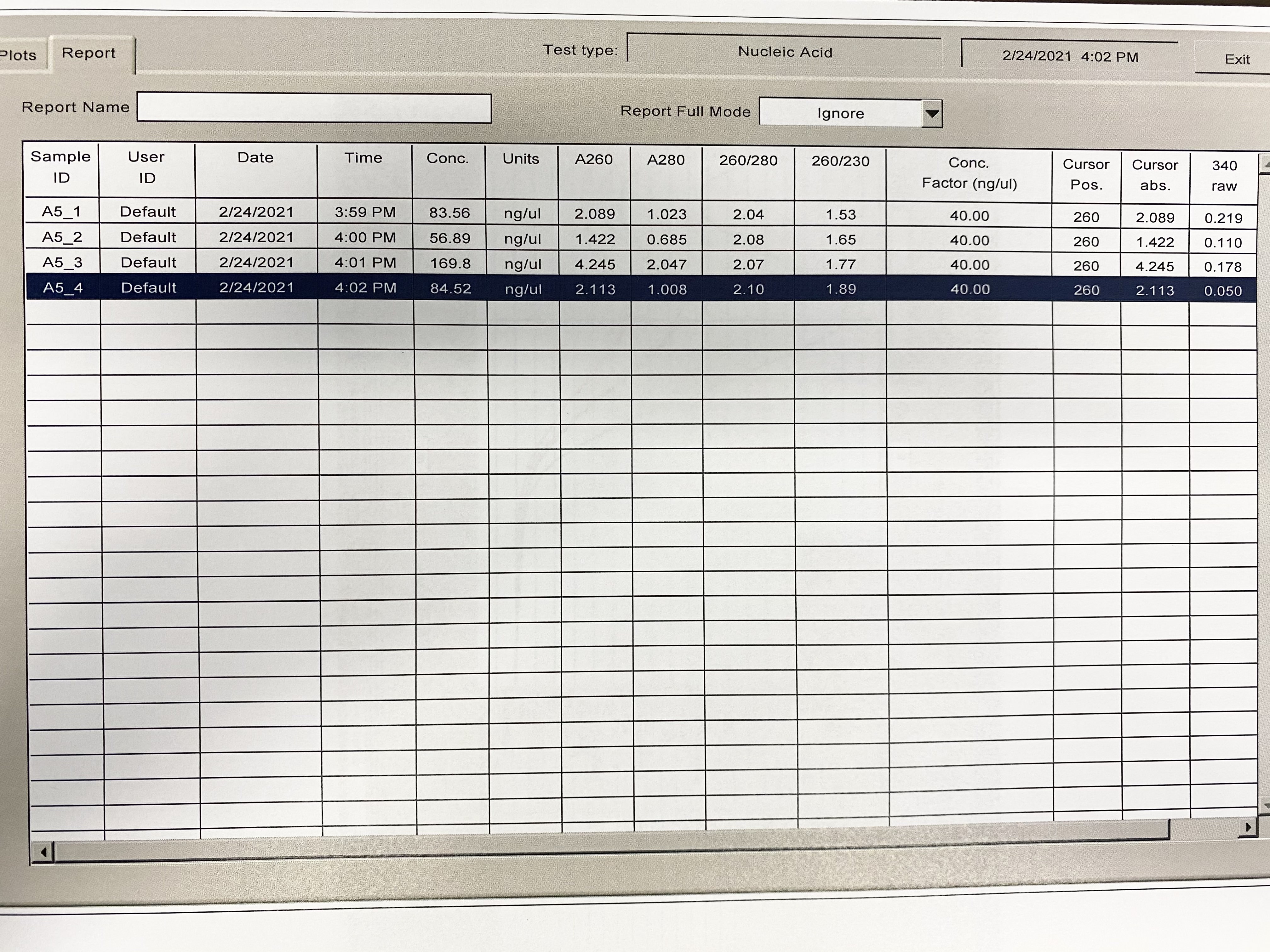
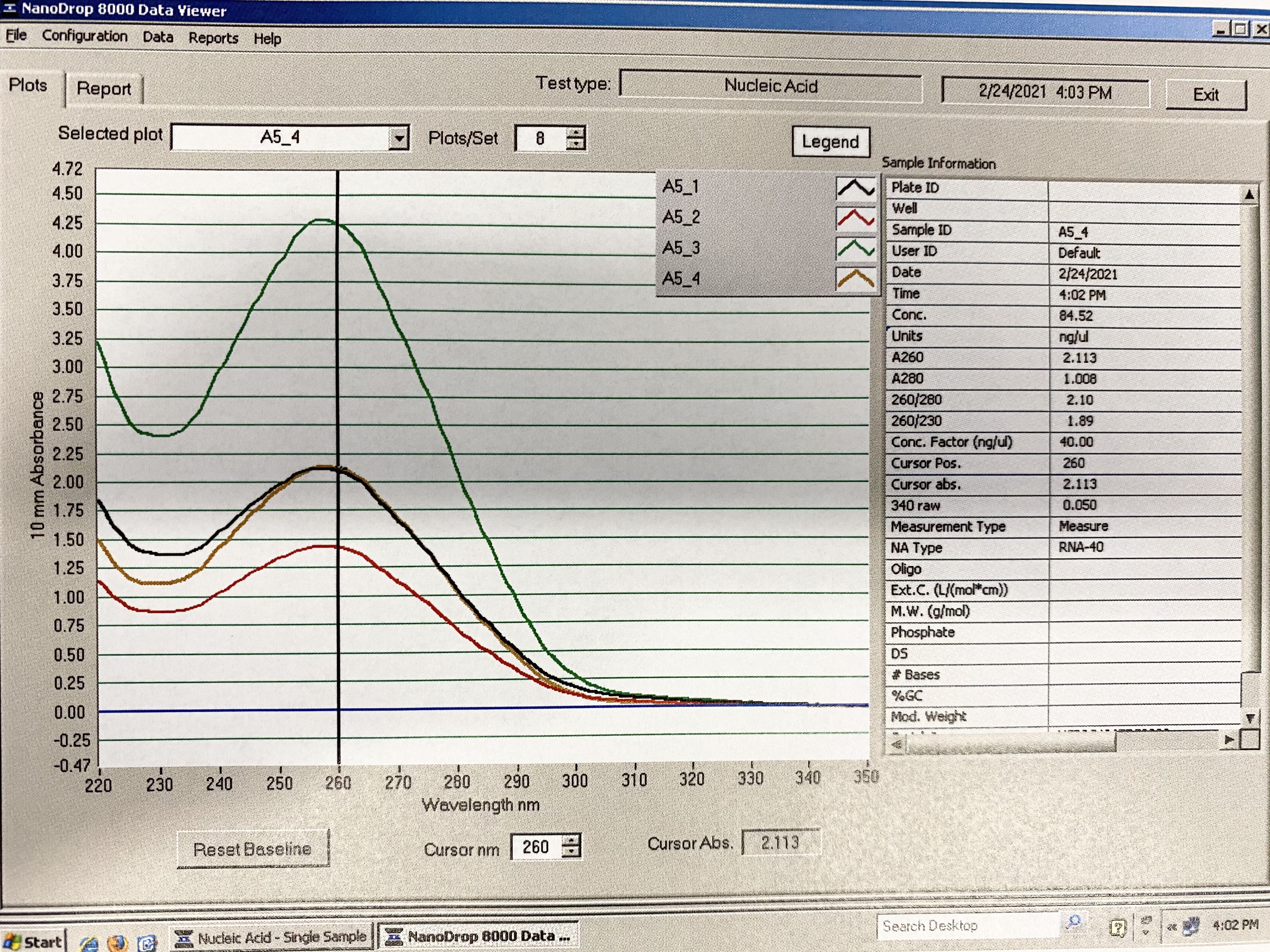
NanoDrop
| Sample | 260/230 | 260/280 |
|---|---|---|
| A5_1 | 1.53 | 2.04 |
| A5_2 | 1.65 | 2.08 |
| A5_3 | 1.77 | 2.07 |
| A5_4 | 1.89 | 2.1 |
Full Results:
Traces:
Gel
- 1% gel run for 1 hour at 80V

- There is some strange streaking/smearing here. It could be an artifact of the gel, or it could be that some of the lipid layer was sucked up and put through the DNA column, and that came out with the DNA
TapeStation
- Followed RNA TapeStation protocol
- Results link