Testing if Cells are Electrocompetent with pTWIN Plasmid and Whether DiNV DNA Gets into Cells
I had made electrocompetent pSPIN-BAC cells but I had not tested to see if they are actually electrocompetent. So I decided to electroporate them with a pTWIN+ plasmid we have in the lab that has Ampicillin resistance and see if they grow on an Amp plate, and use the cells from NEB as a positive control because they should still be electrocompetent (stored in -80).
I also want to test whether DiNV DNA is even getting into the cells by doing DNA extractions from cells just mixed with virus DNA and cells where it was electroporated. I might need to do qPCR to tell but I will start with just regular PCR. I also wanted to test these cells right after electroporation/mixing and with the 1 hour incubation with SOC buffer.
Electroporation
- Prepared 6 1.5mL tubes with SOC buffer
- Thawed pTWIN+ on ice, vortexed and spun down
- Kept sample 20-exo (DiNV DNA) on ice
- Thawed NEB and pSPIN-BAC electrocompetent cells on ice
- Brought up to Chandler lab:
- ice with bacteria and DNA
- pipettes and tips
- bacterial tip waste
- tape
- 20 1.5mL tubes
- tubes with SOC buffer
- Placed 4 SOC buffer tubes in the 37C incubator
- Placed 2 SOC buffer tubes in the 30C incubator
- Placed 5 electroporation EC2 cuvettes on ice
- pSPIN-BAC neg control - tube 1
- Added 25ul pSPIN-BAC cells to a cuvette on ice
- Electroporated at EC2 settings
- Immediately added 975ul of 37C SOC buffer
- Transferred the liquid to a 1.5mL tube labeled “tube 1”
- Placed tube in the 37C shaking incubator 1 hour
- pSPIN-BAC w/pTWIN+ - tube 2
- Added 25ul pSPIN-BAC cells to a tube on ice
- Added 1ul of pTWIN+ (at 200ng/ul, this should be ~200ng)
- Mixed and transferred solution to a cuvette on ice
- Electroporated at EC2 settings
- Immediately added 975ul of 37C SOC buffer
- Transferred the liquid to a 1.5mL tube labeled “tube 2”
- Placed tube in the 37C shaking incubator 1 hour
- NEB electrocompetent cells neg control - tube 3
- Added 25ul NEB cells to a cuvette on ice
- Electroporated at EC2 settings
- Immediately added 975ul of 37C SOC buffer
- Transferred the liquid to a 1.5mL tube labeled “tube 3”
- Placed tube in the 37C shaking incubator 1 hour
- NEB electrocompetent cells w/pTWIN+ - tube 4
- Added 25ul NEB cells to a tube on ice
- Added 1ul of pTWIN+ (at 200ng/ul, this should be ~200ng)
- Mixed and transferred solution to a cuvette on ice
- Electroporated at EC2 settings
- Immediately added 975ul of 37C SOC buffer
- Transferred the liquid to a 1.5mL tube labeled “tube 4”
- Placed tube in the 37C shaking incubator 1 hour
- pSPIN-BAC cells w/DiNV DNA - tubes 5A and 5B
- Added 25ul of pSPIN-BAC cells to a 1.5mL tube on ice
- Added 2.5ul of 20-3xo DNA
- Mixed solution
- Added 975ul of 30C SOC buffer
- Transferred 490ul to a second 1.5mL tube
- Labeled tubes 5A and 5B
- Placed 5B on ice and 5A in the 30C shaking incubator
- pSPIN-BAC cells w/DiNV DNA and electroporation - tubes 6A and 6B
- Added 25ul of pSPIN-BAC cells to a 1.5mL tube on ice
- Added 2.5ul of 20-exo DNA
- Mixed solution and transferred to a cuvette on ice
- Electroporated on EC2 settings
- note here, the electroporated said arc, I kept going but I also re-did this sample kinda
- Added 975ul of 30C SOC buffer
- Transferred 490ul to two 1.5mL tubes
- Labeled tubes 6A and 6B
- Placed 6B on ice and 6A in the 30C shaking incubator
- Ran down to the lab and grabbed DNA sample 22-exo, more SOC buffer, and another tube of pSPIN-BAC cells
- Waited for those to thaw/warm
- pSPIN-BAC cells w/DiNV DNA and electroporation - tubes 7A and 7B
- Added 25ul of pSPIN-BAC cells to a 1.5mL tube on ice
- Added 2.5ul of 22-exo DNA
- Mixed solution and transferred to a cuvette on ice
- Electroporated on EC2 settings - no arching this time
- Added 975ul of 30C SOC buffer
- Transferred 490ul to two 1.5mL tubes
- Labeled tubes 7A and 7B
- Placed 7B on ice and 7A in the 30C shaking incubator
- Left in incubators for 1 hour
Preparing Samples for DNA Extraction
- Took tubes 5B, 6B, and 7B to 4012 and spun for 30 seconds at 13,000rpm
- Removed supernatant
- Resuspended pellets in 300ul LB
- Spun tubes for 30 seconds at 13,000rpm
- Removed supernatant
- Placed pellets of bacteria in the -80
- After incubation for tubes 5A, 6A, and 7A I did the same steps as above, pellet, wash once, then storage at -80
Plating pTWIN Bacteria
- During the 1 hour inubation, I made 12 plates with Ampicillin
- Spread 25ul of stock (100ug/mL) Amp on each of 12 LB plates
- Left the plates in the 37C incubator for ~20 minutes to warm and soak in (the plate got kind of damp after doing this)
- Warmed 2mL of LB buffer to 37C in the incubator
- After the 1 hour incubation, I spun down tube 1-4 at 3,000rpm for 3 minutes
- Removed the supernatant
- Resuspended each pellet in 200ul warmed LB
- Plated each according to the table below:
| plate | volume | tube | sample type |
|---|---|---|---|
| A | 10ul | tube 1 | pSPIN-BAC no DNA |
| B | 40ul | tube 1 | pSPIN-BAC no DNA |
| C | 150ul | tube 1 | pSPIN-BAC no DNA |
| D | 10ul | tube 2 | pSPIN-BAC pTWIN+ |
| E | 40ul | tube 2 | pSPIN-BAC pTWIN+ |
| F | 150ul | tube 2 | pSPIN-BAC pTWIN+ |
| G | 10ul | tube 3 | NEB no DNA |
| H | 40ul | tube 3 | NEB no DNA |
| I | 150ul | tube 3 | NEB no DNA |
| J | 10ul | tube 4 | NEB pTWIN+ |
| K | 40ul | tube 4 | NEB pTWIN+ |
| L | 150ul | tube 4 | NEB pTWIN+ |
- Placed each plate in the 37C incubator upside down overnight
DNA Extractions
- Extracting DNA from bacteria pellets 5A, 6A, 7A, 5B, 6B, 7B
- Took each tube out of the -80
- Added 300ul of chilled/cloudy cell lysis solution to each tube
- Added 0.6ul of RNase A to each tube
- Inverted tubes 25X
- Spun down tubes briefly
- Placed tubes in 37C heat block for 1 hour
- Prepared final tubes with labels and 300ul of 100% isopropanol and prepared fresh 70% ethanol
- After incubation placed tubes on ice
- Added 150ul protein precipitation solution to each tube
- Vortexed tubes and placed on ice for 5 minutes
- Centrifuged tubes for 5 minutes at max speed
- Transferred supernatant (~450ul) to final tubes with isopropanol
- Inverted tubes 50X
- Centrifuged tubes 3 min max speed
- I did not see a pellet here but the amount of bacteria was a lot smaller than other DNA extracts I’ve done
- Discarded supernatant
- Added 300ul of fresh 70% ethanol
- Inverted tubes 2X
- Centrifuged tubes max speed 1 min
- Discarded supernatant
- Let tubes air dry ~45 min on kim wipe
- Resuspended pellet in 20ul DNA hydration solution and let sit on bench overnight
Checking Plates
- Kistie had taken the plates out of the 37C incubator at ~9am on 20240220 morning
- There was growth on every plate, and I think this is because of two reasons. When adding the bacteria to the plate, they are on LB that doesn’t have antibiotics and they can grow on that for a while (especially the 150ul samples). Also that when I added the anitbiotic and let it soak in in the 37C incubator, the plates got really wet. And I wasn’t sure how to dry them and I think the bacteria moved around a lot on that wetness. But it still looks like to me that the bacteria that did not get the pTWIN+ has much less growth than the bacteria that did. And it didn’t matter which electrocompetent cell I used
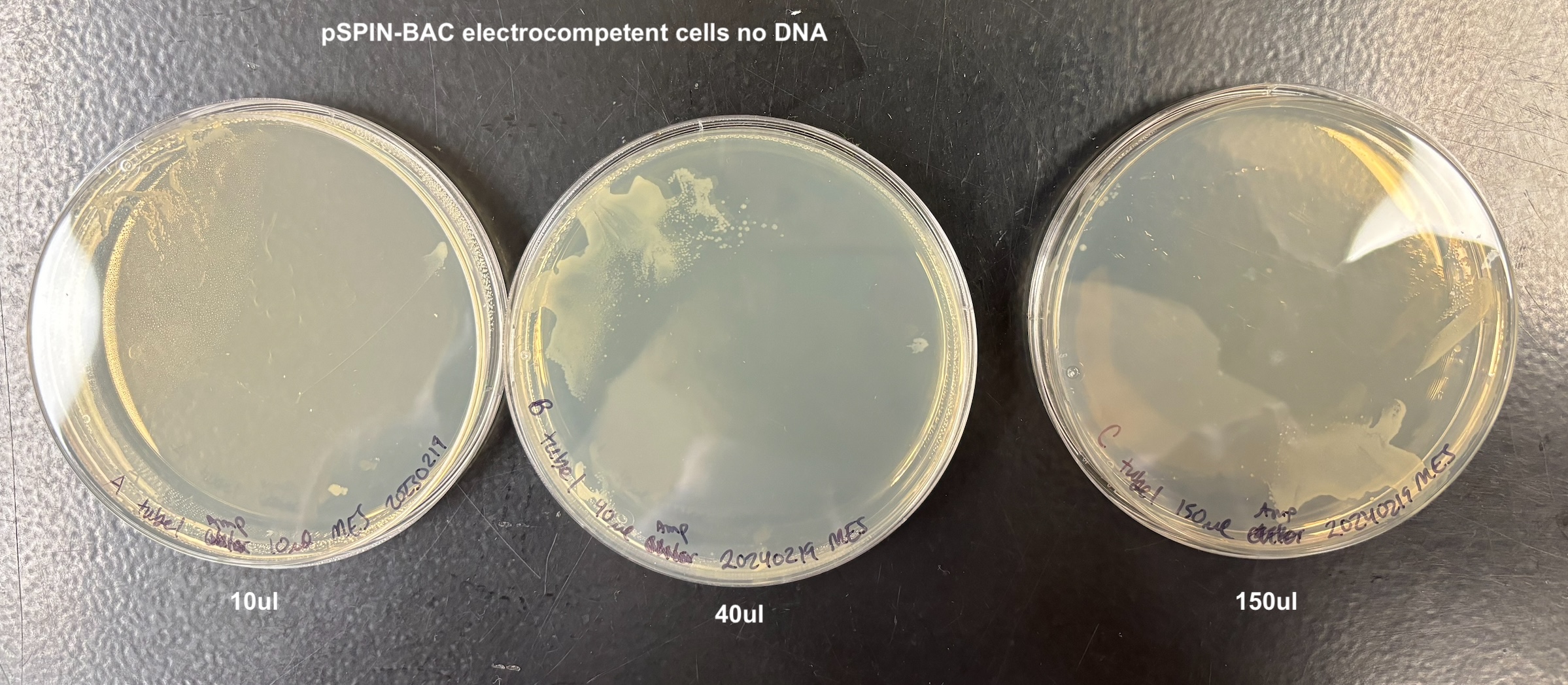
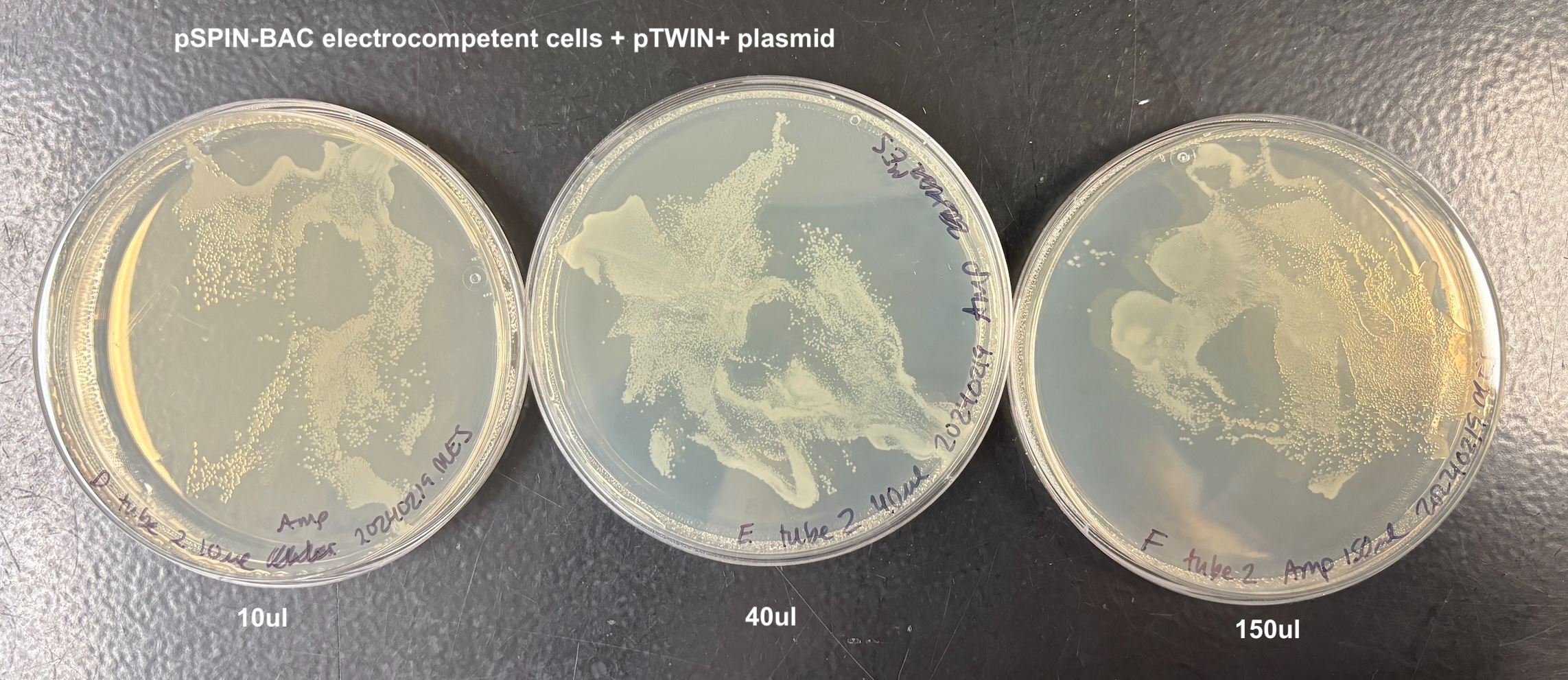
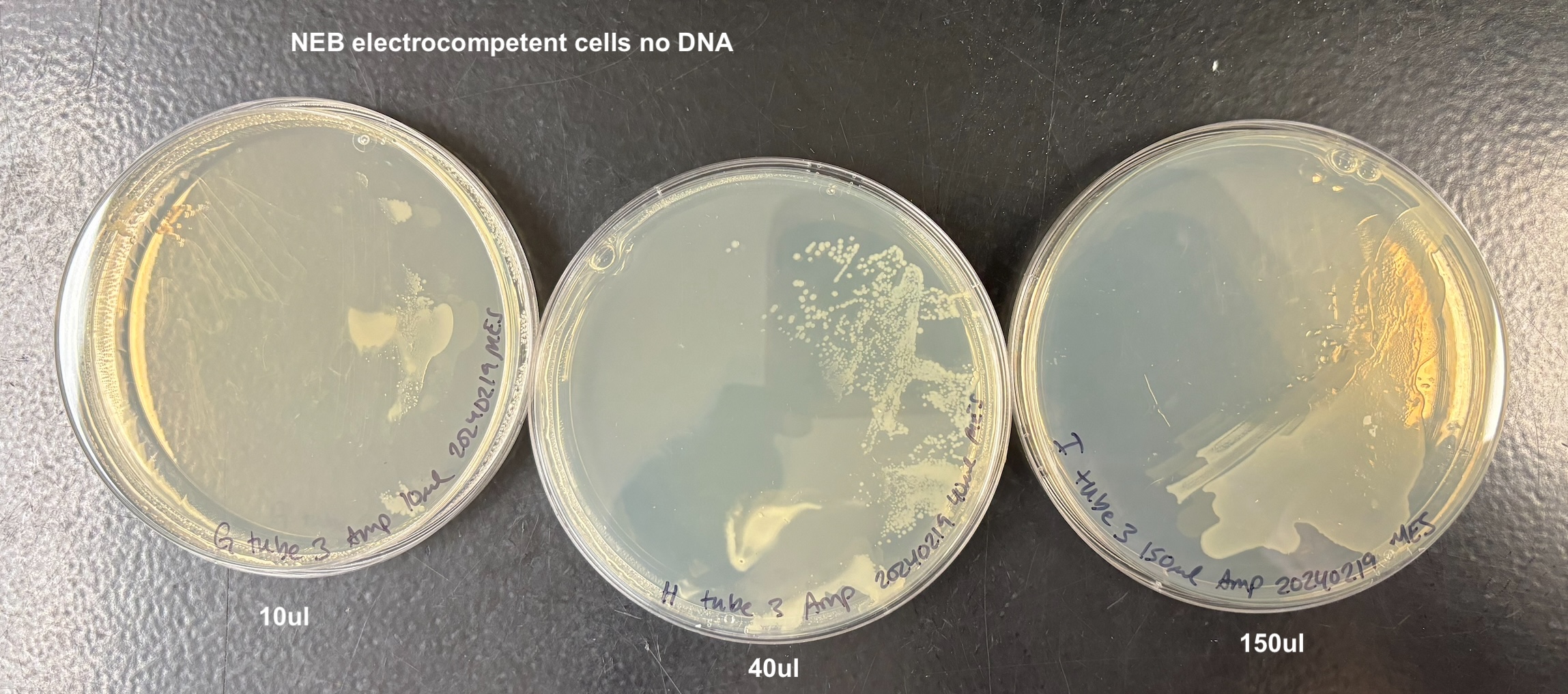
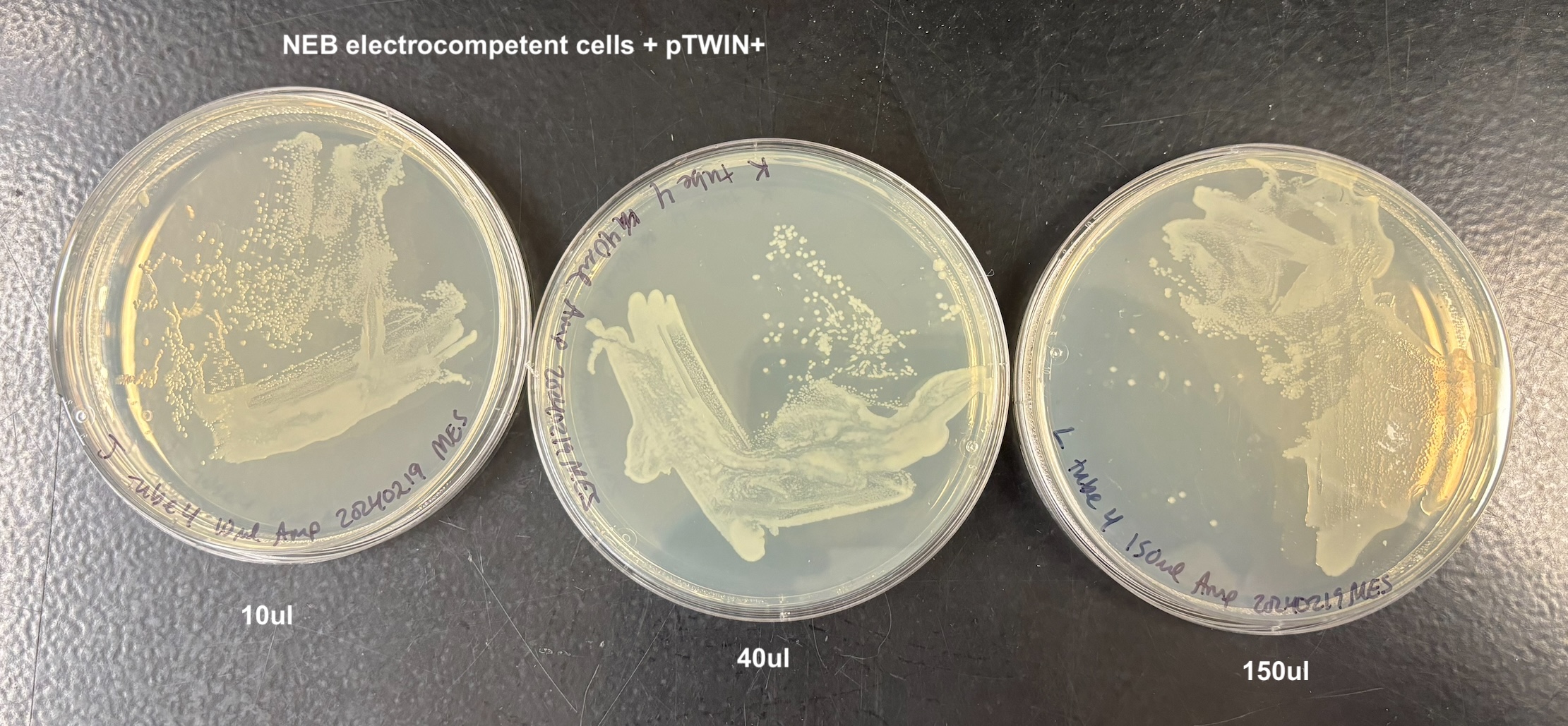
From this I concluded that the pSPIN-BAC electrocompetent cells I made are electrocompetent.
PCRs on DNA Extracts
- I want to test all of the DNA samples for if they have retained DiNV DNA with PCR (I might have to do qPCR as well)
- 4 PCRs were run on the samples, and the process followed the general PCR protocol completely. Master mix volumes are listed here:
| reagent | p47 | 16S | lef 9 | lef 4 |
|---|---|---|---|---|
| GoTaq | 42.5ul | 42.5ul | 42.5ul | 42.5ul |
| F primer | 2.125ul | 2.125ul | 2.125ul | 2.125ul |
| R primer | 2.125ul | 2.125ul | 2.125ul | 2.125ul |
| molecular grade water | 29.75ul | 29.75ul | 29.75ul | 29.75ul |
- All PCR programs were run for 35 cycles except for the 16S program which ran for 30 cycles, and program information can be found here
- A 1% gel was run at 90V for 45 minutes to resolve the bands:

All samples amplified, so either the cells who didn’t get electroporated took up the DNA, or that the cells were not washed enough to remove any DNA on the outside of the cells. I will have to try this again.