Using the Band 1, Band 2, and Pool of 1 and 2 Harvest Samples From DiNV Purification for Hirt DNA Extraction for Viral DNA
Based on the degraded DNA from the clarification pellet, I thought I would try extracting from what we think is purified virus with small amounts of fly DNA. I took 50ul sample from the purified band 1 (prefilter), 50ul from the purified band 2 (prefilter), and from the pool of harvest 1 and 2 supernatant (this has been kept at 4 C so expectation is that fly DNA is degraded here). Those are samples 7, 8, and 9 respectively. Sample information is here.
Lysis and Chromosomal DNA Precipitation
- Placed small centrifuge in fridge
- All processes were done in the fume hood
- All samples were in 50ul liquid, so I added 100ul of 50mM Tris HCl, 10mM EDTA to each
- Prepared 5mg/mL RNase A
- 5ul molecular grade water
- 5ul 10mg/mL RNase A
- Vortexed and spun down to mix
- Added 3ul of 5mg/mL RNase A to each sample
- Added 150ul of 1.2% SDS to each tube
- Inverted tubes, gently resuspended pellet with clipped pipette tips to mix
- Incubated tubes on bench for ~20 minutes to lyse/digest all cells/virus
- It was hard to tell is anything happened here because all samples were basically a clear liquid to begin with, but I gently mixed a few times
- Precipitated chromosomal DNA and cellular debris by added 210ul of 3M CsCl, 1M potassium acetate, 0.67M acetic acid
- A white precipitate immediately started forming in the tubes, I pipette mixed the solution with clipped pipette tips
- Immediately placed tubes on ice for 30 minutes incubation
- Centrifuged tubes for 15 minutes at 16,000rcf at 4 degrees C
- During this time I made fresh 80% and 100% ethanol and put them in the -20 freezer to cool
- Pipetted off supernatant into new 1.5mL tubes with clipped pipette tips
- Centrifuged new tubes for 15 minutes at 16,000rcf at 4 degrees C
- There was basically no new precipitate here that pelleted
- Moved supernatant into new 1.5mL tubes
- 7: 440ul
- 8: 490ul
- 9: 490ul
Phenol Chloroform Extraction
- Still in the fume hood
- Added equal volume (500ul) of cold phenol-chlorofomr-isoamy-alcohol to the tubes (Note!! phenol-chloroform-isoamy-alcohol is in two phases, I think you are supposed to use the bottom layer for this, see x)
- 7: 440ul
- 8: 490ul
- 9: 490ul
- Mixed tubes up and down and then put them on an orbital mixer in the hood for 10 minutes
- Centrifuged tubes at 16,000rcf at room temp for 15 minutes
- Looked for phase separation
- Phase separation looked great, there were two distinct layers
- Transferred top aqueous phase to new final labeled tubes (did not use clipped tips here for better pipetting )
- I did not try to get absolutely everything, I did not want to suck up both phases
- 7: 440ul
- 8: 490ul
- 9: 490ul
- Added 0.1X tube liquid volume of new 3M NaOAc to each tube
- 7: 44ul
- 8: 49ul
- 9: 49ul
- Added 900ul of cold 100% ethanol to each tube
- Inverted tubes to mix
- I did not see anything that looked like DNA
- Placed tubes in the -20 overnight
DNA Precipitation 20231028
- Took tubes out of the freezer and centrifuged tubes at max speed at 4C for 30 minutes
- Tubes had not frozen in the freezer
- Maybe tube 9 had a pellet, all others looked like nothing
- Pipetted off supernatant
- Added 500ul of cold fresh 80% ethanol and inverted twice to mix
- Centrifuged tubes at max speed at 4C for 30 minutes
- Removed all ethanol
- Let tubes dry ~60 minutes upside-down on the bench
- Resuspended pellets in 25ul of 10mM tris HCl and let resuspend in the 4C until Monday
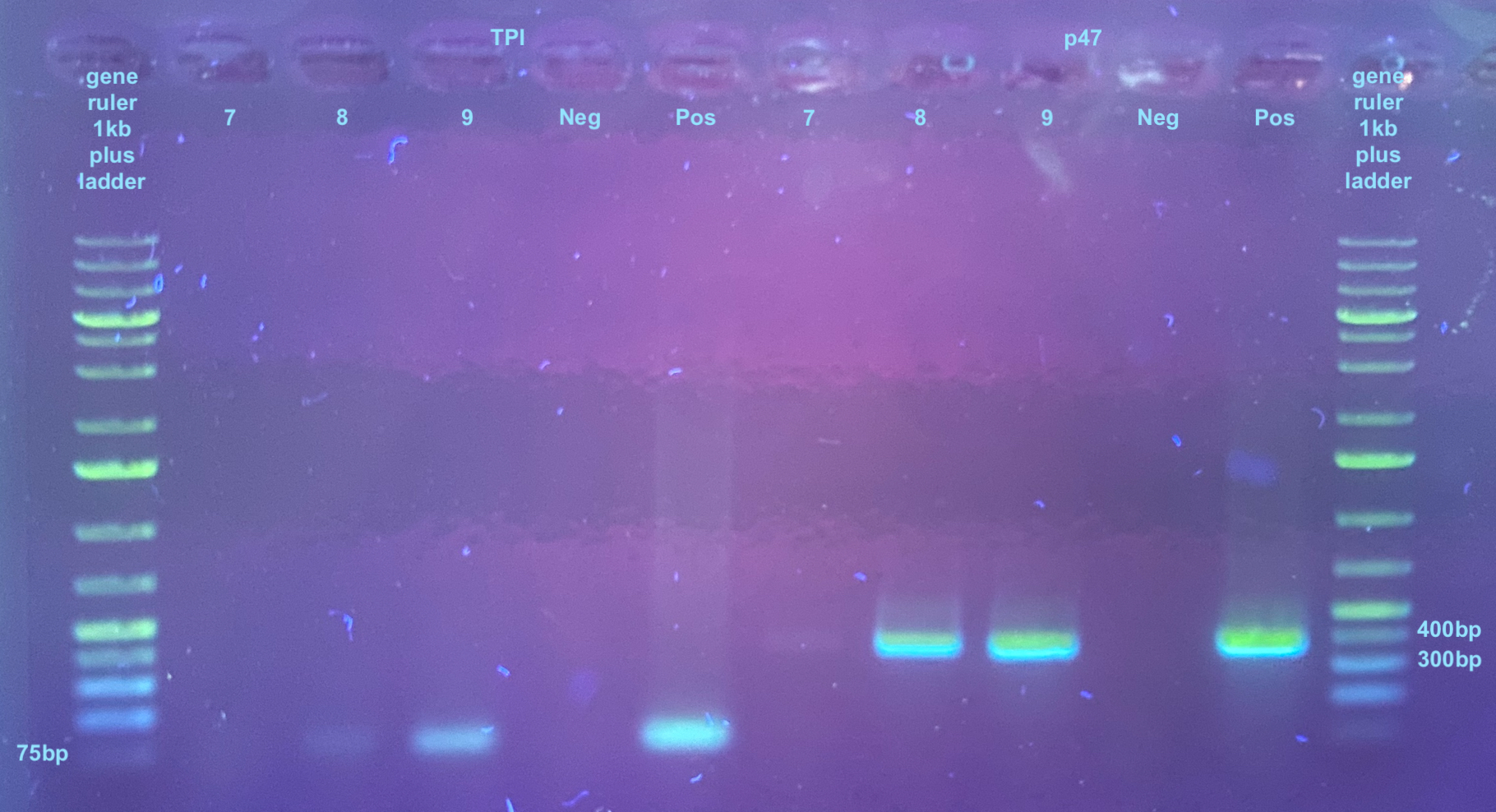
TPI and p47 PCRs and Gels 20231031
- There are 3 samples
- Include positive and negative control
- Everything was thawed and kept on ice, and vortexed and spun down before use
- TPI
- 27.5ul GoTaq
- 1.375ul TPI F
- 1.375ul TPI R
- 19.25ul molecular grade water
- p47
- 27.5ul GoTaq
- 1.375ul p47 F
- 1.375ul p47 R
- 19.25ul molecular grade water
- Vortexed and spun down mixes
- Added 9ul of mixes to strip tubes
- Added 1ul of diluted DNA to tubes
- Added 1ul of water for the negative control and 1ul known virus positive DNA for the positive control
- Vortexed and spun down strip tubes
- Placed tubes in PCR programs, 30 cycles for each. PCR program information can be found here
- Afterwards I did a broad range DNA qubit on each sample and they were all too low to read on with these reagents. This means each sample must be less than 2ng/ul concentration, which is far lower than the amount I’d need for electroporation…
All samples were run on a 1% gel : small rectangle 30mL 1X TAE and 0.3g agarose, .5ul of Midori stain