Third Test of the Hirt Method for Viral DNA Extraction With Phenol Chloroform Purification on DV-2 Cells Infected with DiNV, and p47 and RPL11 PCRs on Extracted DNA
Second extraction test did not go well because the DNA was too contaminated with something to get a PCR to work. I did a nanodrop on the extracts and the 260/280 ration was high (between 1.85 and 2.13) which indicates that the solution is basic. What I changed this time is that I centrifuged the samples twice after the chromosomal precipitation, I did not use phase lock tubes, I did the incubation for DNA precipitation in the -20 overnight, and I made new NaOAc.
New 3M NaOAc
- Making 50mL
- 12.3g sodium acetate
- Dissolved in 35mL of molec grade water (took a while to dissolve, lots of mixing)
- Checked pH with test strips, want it to be between 4.8 and 5.2
- Original pH was between 6 and 7, way too high
- I did many “titrations” of adding glacial acetic acid, but it came out to be about 1.2mL of glacial acetic acid added to get the pH to about 5 (test strips aren’t super accurate)
- Then I increased the volume to 50ul with molec grade water
Lysis and Chromosomal DNA Precipitation
- Placed small centrifuge in fridge at noon
- Thawed 2 samples from Kent and a cell control Dv-1 sample to use as a sort of control for the extraction
- Samples named by the volume used:
- 1.5-2 : DiNV GS 20181123 Dv-1-2 day 10 lysate
- 0.75 : DiNV GS 20181123 Dv-1-3 day 10 lysate
- 0.5-C : DiNV GS 20181123 Dv-1-CC day 10 lysate
- Mixed the tubes before transferring samples to 1.5mL tubes
- Used clipped pipette tips to transfer volume to new 1.5mL tubes
- Centrifuged tubes at 4C 1000rcf for 3 minutes
- Each tube had a clear pellet
- Removed supernatant from pellet
- Added 1.5mL 1X PBS
- Centrifuged tubes at 4C 1000rcf for 3 minutes
- Removed supernatant from pellet
- Resuspended pellet in 147ul 50mM Tris HCl, 10mM EDTA and 3ul of 5mg/mL RNase A
- Added 150ul of 1.2% SDS to each tube
- Inverted tubes, gently resuspended pellet with clipped pipette tips to mix and spun down
- Incubated tubes on bench for 10 minutes to lyse all cells, waited until I couldn’t see any cell chunks anymore
- Precipitated chromosomal DNA and cellular debris by added 210ul of 3M CsCl, 1M potassium acetate, 0.67M acetic acid
- A white precipitate immediately started forming in the tubes
- Immediately placed tubes on ice for 25 minutes incubation (this was increased from 15 minutes)
- Centrifuged tubes for 15 minutes at 16,000rcf at 4 degrees C
- Pipetted off supernatant into new 1.5mL tubes with clipped pipette tips
- Centrifuged tubes for 15 minutes at 16,000rcf at 4 degrees C
- There was basically no new precipitate here that pelleted
- Moved supernatant into new 1.5mL tubes ~475ul
Phenol Chloroform Extraction
- Took tubes and setup to the hood
- Added equal volume (475ul) of cold phenol-chlorofomr-isoamy-alcohol to the tubes
- Mixed tubes up and down and then put them on an orbital mixer in the hood for 10 minutes
- Centrifuged tubes at 16,000rcf at room temp for 15 minutes
- Looked for phase separation
- Phase separation was “perfect” in each tube
- Transferred top aqueous phase to new final labeled tubes
- I did not try to get absolutely everything, I did not want to suck up both phases
- 400ul, 550ul, and 400ul were sucked up
- Added 0.1X tube liquid volume (either 40ul or 55ul) of new 3M NaOAc to each tube
- Added 900ul of cold 100% ethanol
- Inverted tubes to mix
- Placed tubes in the -20 overnight
DNA Precipitation 20220902
- Centrifuged tubes at max speed at 4C for 30 minutes
- Tubes had not frozen in the freezer
- A small pellet was seen in all tubes after this
- Pipetted off supernatant
- Added 500ul of cold fresh 80% ethanol
- Centrifuged tubes at max speed at 4C for 30 minutes
- Removed all ethanol
- Let tubes dry ~60 minutes upside-down on the bench
- After drying I couldn’t see the pellet anymore…
- Resuspended pellets in 25ul of molecular grade water and let sit on the bench for a few hours
- Qubit that afternoon:
- 1.5-2 : 63.7ng/ul
- 0.75 : 35.5ng/ul
- 0.5-C : 130ng/ul
- The yield is nice here, but in theory the cell control sample shouldn’t have any DNA in this extraction because there isn’t any virus. So this is kind of sad. I expected that I could never get rid of all of the Dv-1 DNA but this is a lot.
p47 and RPL11 PCRs 20220906
- p47
- So I have 3 samples, plus 1 for a positive control, and plus 1 for a negative control
- Made master mix on ice:
- 5ul GoTaq * 5.5 = 27.5ul
- 0.25ul p47_F * 5.5 = 1.375ul
- 0.25ul p47_R * 5.5 = 1.375ul
- 3.5ul molecular grade water * 5.5 = 19.25ul
- Vortexed and spun down master mix
- Added 9ul master mix to 4 strip tubes
- Added 1ul of DNA sample to appropriate tubes
- Added 1ul of 3mL HMW extraction to the positive control tube
- Added 1ul of molecular grade water to the negative control tube
- Vortexed and spun down strip tubes
- Placed tubes in the p47 PCR program
- RPL11
- Using the same sample scheme as above
- Made master mix on ice:
- 5ul GoTaq * 5.5 = 27.5ul
- 0.25ul p47_F * 5.5 = 1.375ul
- 0.25ul p47_R * 5.5 = 1.375ul
- 3.5ul molecular grade water * 5.5 = 19.25ul
- Vortexed and spun down master mix
- Added 9ul master mix to 4 strip tubes
- Added 1ul of DNA sample to appropriate tubes
- Added 1ul of 3mL HMW extraction to the positive control tube
- Added 1ul of molecular grade water to the negative control tube
- Vortexed and spun down strip tubes
- Placed tubes in the RPL11 PCR program
- Ran a 2% gel for 35 minutes at 90 volts after the PCR programs were done:
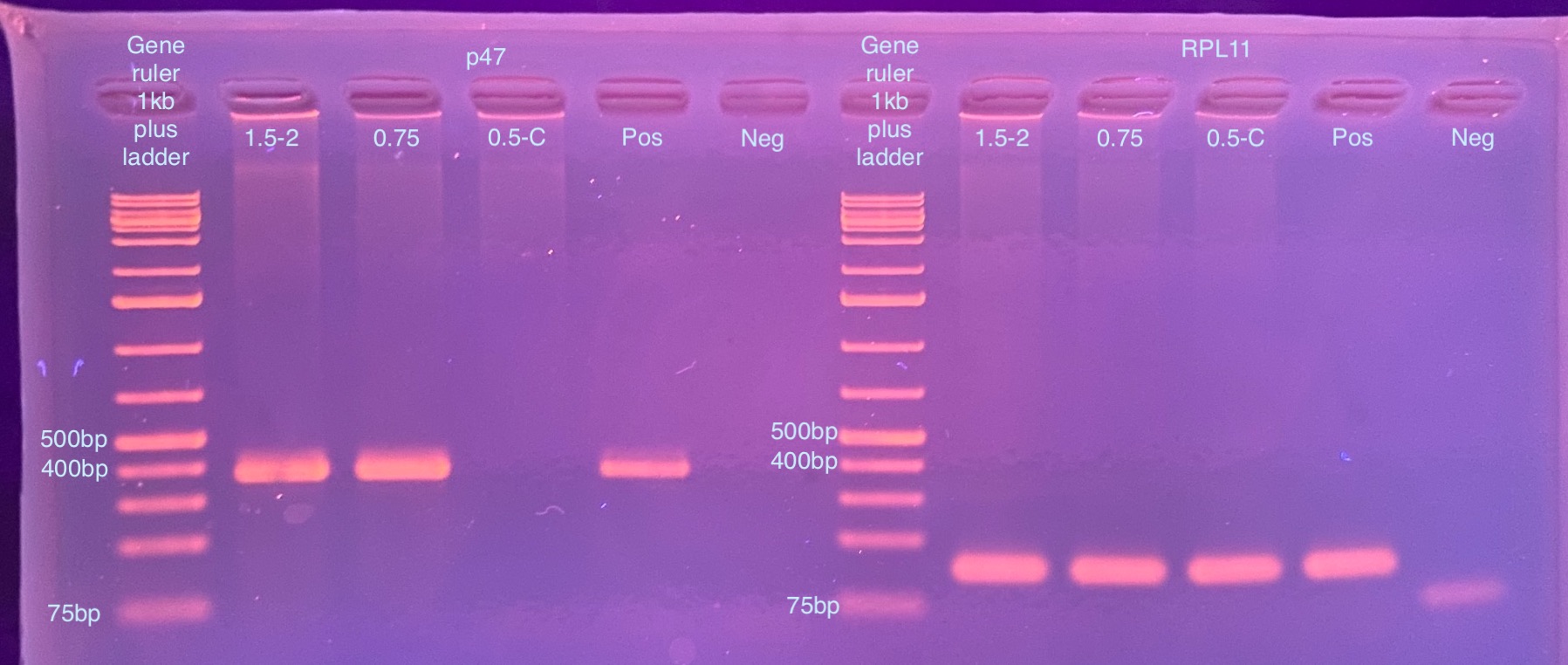
Clearly there is still Dv-1 DNA in all of these, I’m not sure what more I can do to minimize that…