First Attempt at Running a 16 Hour Gel on HMW DNA Samples, Cutting out a “150kb” Region, and Gel Extracting
Notes
- Running the gel 15-16hrs overnight because this way I’m able to have time right after it’s done to do a gel extraction, and the ~15 hour gel looked ok in terms of resolution the last I tried it
- Using a 0.8% gel and running at 40V because this keeps all the DNA on the gel, and a lower gel percentage is good for the extraction
- Using the QIAquick Gel Extraction Kit
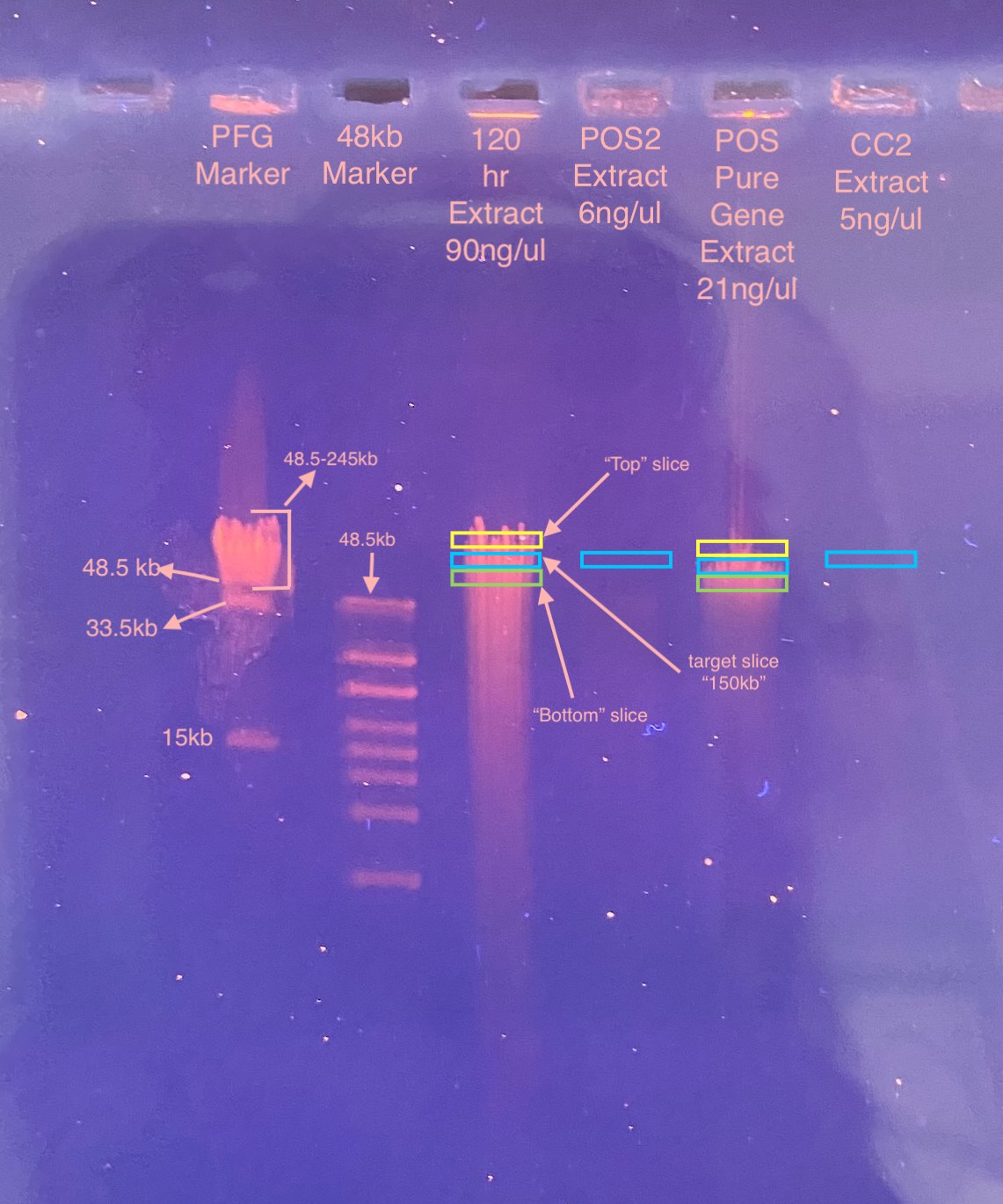
- Decided to run my best sample on the gel (90ng/ul), as well as an ok POS2 sample, a cell control sample, and the POS PureGene kit sample on the gel. I wanted some kind of extraction control (CC2) for doing the DiNV specific PCR on. And the PureGene sample may not have HMW DNA in the right size but I wanted to try it
- When slicing the gel, because we don’t have any separation of the bands in the PFG marker between 48.5-245kb, I was really guessing on where the “150kb” size would be. I decided to cut out a slice where I guessed it would be, and then also a slice above and below that region to see if I could amplify DiNV in those sizes too
20211115 Loading and Running Gel
- .8% gel mix:
- 180mL 1X TAE
- 1.4g agarose
- Microwaved for ~4min
- Pipetted 1mL into a 1.5mL tube and saved on the heat block at 65 degrees C make sure the heat block is on and running
- Made sure the large tray had well sealed tape barriers
- Let the gel liquid cool for ~10 min until pouring into the tray and placing in the combs
- Gel cooled in ~20 min or less
- Added 1 round of PFG ladder to the 3rd left most well (there looked like there were some imperfections near the edge of the gel)
- Let the 1mL saved gel cool for ~3 min outside of the heat block
- Filled the PFG well with cooled gel liquid to the top of the well to seal the rounds in
- Waited ~3 minutes for the wells to cool
- Made up 48kb ladder:
- 1.2ul ladder
- 6ul loading dye
- 28.8ul molecular grade water
- Placed the gel tray into the box after removing the tape
- Added 16ul 48kb ladder to the well right next to the PGF well
- Mixed 5ul 120hr sample with 1ul loading dye and added into the 3rd well
- Mixed 7ul of POS2, POSPG, and CC2, each with a respective ul of loading dye and added in that order to the gel
- Set the timer for 16 hours at 40 volts and started it at ~5pm
20211116 Imaging, Cutting, and Gel Extraction
- Stopped the gel at ~9am
- Sliced the gel in half and put the half with samples in the EtBr bath for 1 hr
- Prepped for imaging and cutting the gel: made tubes for the gel pieces, got glasses, lab coat, gloves, and face shield ready, cleaned a razor blade with ethanol
- First, imaged the gel and saw that POS2 and CC2 were basically too faint to see. These had the lowest DNA quants but I was hoping I’d see them. Also the PFG marker looked more scrunched than I thought it would
- I decided to still try to cut “150kb” sizes from the two samples I couldn’t see, but only cut the top and bottom slices from the 120hr and POSPG
- It was hard to slice out the right section of the gel, the bottom (where the DNA was) didn’t super want to come out of the gel. I did not cut off any excess gel without DNA because I was worried about having the UV on too long on the DNA
- Then I weighed each tube to get the weight of the gel piece. A tube without any gel in it was 0.9886g
| Sample name | Tube with gel weight (g) | gel weight (mg) | 3 volumes of gel (ul) |
|---|---|---|---|
| 120hr | 1.018 | 29.4 | 88.2 |
| POS2 | 1.028 | 39.4 | 118.2 |
| POSPG | 1.025 | 36.4 | 109.2 |
| CC2 | 1.033 | 44.4 | 133.2 |
| 120hr Top | 1.04 | 51.4 | 154.2 |
| POSPG Top | 1.016 | 27.4 | 82.2 |
| 120hr Bottom | 1.022 | 33.4 | 100.2 |
| POSPB Bottom | 1.012 | 23.4 | 70.2 |
- Added “3 volumes” of the gel to each tube with Buffer QG (4th column in table above)
- Note here that the QG buffer should be yellow to have the right pH. The buffer in the bottle looked a little orange, but in the tube looked nicely yellow, so I did not add and NaOAc to adjust the pH


- Note here that the QG buffer should be yellow to have the right pH. The buffer in the bottle looked a little orange, but in the tube looked nicely yellow, so I did not add and NaOAc to adjust the pH
- Tubes were spun down and placed in a heat block for 10 minutes at 50 degrees C
- Tubes were vortexed and spun down once during the incubation
- Tube color was still yellow in my opinion at this point
- Added 1 gel volume (column 3 in table above, but in ul) of 100% isopropanol to each tube and inverted to mix
- Set up spin columns for each sample
- Added total volume to the spin columns with clipped pipette tips (~200ul)
- Centrifuged columns 1 min, 17,900g
- Discarded flow through
- Added 500ul of buffer QG to each column. Kit protocol says: “Recommended: Add 0.5 ml of Buffer QG to QIAquick column and centrifuge for 1 min. This step will remove all traces of agarose.” The buffer QG was pretty orange at this volume, and may have caused me problems…
- Centrifuged columns 1 min, 17,900g
- Discarded flow through
- Added 750ul buffer PE to each column
- Centrifuged columns 1 min, 17,900g
- Discarded flow through
- Centrifuged columns 1 min, 17,900g “dry”
- Transferred columns to final 1.5mL tubes
- Added 50ul buffer EB to the columns
- Let tubes sit for 5 minutes
- Centrifuged columns 1 min, 17,900g
- Removed columns and stored tubes
- Qubitted tubes immediately, all samples except for the 120hr slices had too low to read DNA
- 120: 0.11ng/ul
- 120T: 0.18ng/ul
- 120B: 0.212ng/ul
I’m hoping here the issue was that the buffer QG was not the right pH for DNA binding. I will try the overnight gel again with more input DNA from the 120hr extract, probably trying two different lanes. I don’t think the amount of DNA input is an issue because that sample is really concentrated in DNA, but I don’t know. I will try 15ul which should be over 1ug of DNA. The low yield samples basically didn’t show up in the gel, there is resolution lost running it so long so that makes sense. Another issue could be that the DNA was too large to elute from the spin column, the size I want is out of the range of the kit. But I just expected the DNA to shear not to not come out. I can adjust the elution incubation time and warm the elution buffer to help the DNA release from the column. I will try this again!