DiNV Gene p47 PCR and Gel of HMW Extracts and Supernatant from 1st Successful Extraction Attempt, Two Tries on the PCR
Notes
- Want to see if the HMW extraction yielded any virus DNA. Even though the amount of DNA I got was low, it should be usable for a PCR to check that the samples I’m using have the virus.
- I also wanted to check if the supernatant from the precipitation would amplify for DiNV, because that would be a confirmation of DNA in the supernatant.
- Because of the low concentration of the extracted DNA, I adjusted the standard PCR recipe to use the volume for water as DNA. Instead of 1ul DNA and 6.4ul water per sample, I used 7.4ul DNA from every sample except the negative and positive controls. With sample DNA concentrations 0.2ng/ul-0.4ng/ul that would be about 3ng total DNA going into the PCR. PCRs I have done before I usually start with 10ng input DNA.
- The negative control was molecular grade water
- The positive control was from Kent, I’m not sure what it is but he gave it to me as a positive control, I used 1ul of that sample
- I did not have diluted primers, so first I had to dilute primers. I used p47_F/R_q for this reaction. After preparing it I realized that the “q” may mean qPCR. These should still work for regular PCR, but I made an aliquot of 10uM p47_F/R primers as well for the future
Primer Dilution
- Stock primers should be at 100uM, want to make working stocks at 10uM
- Thawed stock primers on ice, vortexed and spun down
- P47_F_q
- 90ul molecular grade water
- 10ul 100uM primer
- P47_R_q
- 90ul molecular grade water
- 10ul 100uM primer
- P47_F
- 90ul molecular grade water
- 10ul 100uM primer
- P47_R
- 90ul molecular grade water
- 10ul 100uM primer
- Vortexed and spun down 10uM primers
- Kept primers on ice for use
- Stock primers went into stock primer boxes
- My aliquots are in a new “Maggie PCR” box on Kent’s shelf in the -20 close to Jessie’s desk
PCR Setup
- Thawed NEB 10X reaction buffer, NEB One Taq Aliquot, 2mM dntps on ice
- Prepared master mix with the calculations on the lab drive for a 10ul total reaction volume
- Decided to do each extraction product twice (6), as well as the supernatant from the precipitation (2), and a negative and positive control (2), for a total of 8 samples + error = 8.5 = n
- Master mix on ice:
- 1ul 10X buffer * 8.5 = 8.5ul
- 1ul 2mM dntps * 8.5 = 8.5ul
- 0.25ul p47_F_q * 8.5 = 2.125ul
- 0.25 P47_R_q * 8.5 = 2.125ul
- 0.1ul Taq * 8.5 = 0.85ul
- Vortex and spin down master mix, keep on ice
- Set up a strip tube for the samples on ice
- Sample order: POS1, POS2, CC1, CC2, S1, S2, N, P
- POS is the DiNV positive cells I extracted, CC is the cell control cells I extracted, S is the supernatant from the ethanol precipitation, N is the negative control, and P is the positive control
- Added 7.4ul of DNA from samples to their respective strip tubes for POS, CC, and S tubes
- Added 7.5ul molecular grade water to the N tube
- Added 6.4ul molecular grade water to the P tube
- Added 1ul of the positive control sample to the P tube
- Added 2.6ul of master mix to every strip tube
- Vortexed and spun down the strip tubes
PCR Conditions
- Tm of both primers is 60.4 degrees C, so I decided to do an annealing temp of 55 degrees C. I didn’t know if there is a PCR program made already, there probably is but I don’t know where. Hopefully the program I made works
- Followed roughly the suggestions from NEB about the cycling conditions
- 95 degrees C 30 seconds
- 95 degrees C 30 seconds
- 55 degrees 30 seconds
- 68 degrees C 30 seconds
- 68 degrees C 5 minutes
- 10 degree C hold
- bold conditions are cycled 30 times
- I named this protocol p47 and made a new folder for myself: Maggie
- Put my tubes in the thermocycler and started it
- Run time ~1 hour 21 minutes
- After the PCR, the tubes were placed in the 4 degree fridge overnight
Gel 20211020
- 1% was already made, just had to warm up and pour into the small gel, used about half of what was from the 100mL pre-made
- Used 3ul of NEB 100bp ladder
- Used 1ul of Qiagen 5X loading dye for the samples
- Used 4ul of each sample
- Ran gel for ~1 hour at 100V
- After done, placed gel in the ETBr bath rotating for ~20 minutes
Gel did not come out well at all. Ladder was barely visible, and there were no bands in any of the samples. Was not able to get a photo that even showed the ladder
So, what to do?
- Next time, pour a way thinner gel. I think it was twice as thick as it needed to be
- Let the gel soak in the ETBr for 30 minutes next time
- Look up the PCR conditions, see if anyone else has a protocol, because something definitely went wrong
I noticed that I had probably used the wrong Taq/wrong PCR mix page. We only have tubes of OneTaq from NEB right now, which is a PCR mix already. I had been following the NEB regular Taq mix sheet. This resulted in me having basically no Taq in the reaction above, which is my best guess to why it didn’t work. I also found a p47 PCR protocol from Kaitlin that has different PCR cycle times than what I used. I am going to try those.
2nd Try p47 PCR 20211020
- I looked up the NEB OneTaq Mix instructions for making the PCR mix and followed those for a 12.5ul reaction volume. This is a 2X mix, so half of the PCR reaction should be this mix. Product number from NEB is M0482
- I also used the regular p47 F and R primers this time, instead of the ones that say “q”
- I’m doing the same sample scheme as above: 8 samples, n = 8.5
- Master Mix
- 0.25ul 10uM p47_F * 8.5 = 2.125ul
- 0.25ul 10uM P47_R * 8.5 = 2.125ul
- 6.25ul 2X OneTaq Mix * 8.5 = 53.125ul
- Vortexed and spun down master mix, kept on ice
- Added 5.75ul DNA to their respective strip tubes for POS, CC, and S
- Added 5.75ul molecular grade water to the N tube
- Added 4.75ul molecular grade water to the P tube
- Added 1ul positive control sample to the P tube
- Added 6.75ul master mix to each tube
- Vortexed and spun down tubes until no bubbles
New PCR cycling conditions
- 94 degrees C 5 minutes
- 94 degrees C 45 seconds
- 55 degrees C 1 minute
- 68 degrees C 1.5 minutes
- 68 degrees C 5 minutes
- Hold at 12 degrees C
- bold text is cycled through 30 times
Program runs for 2 hours and 20 minutes, likely not enough time for me to do the gel before journal club. I will have to try tomorrow if possible.
Gel 20211021
- Kistie set up a 1% gel for me
- Ran at 100V for ~50 minutes
- Soaked in ETBr for 25 minutes
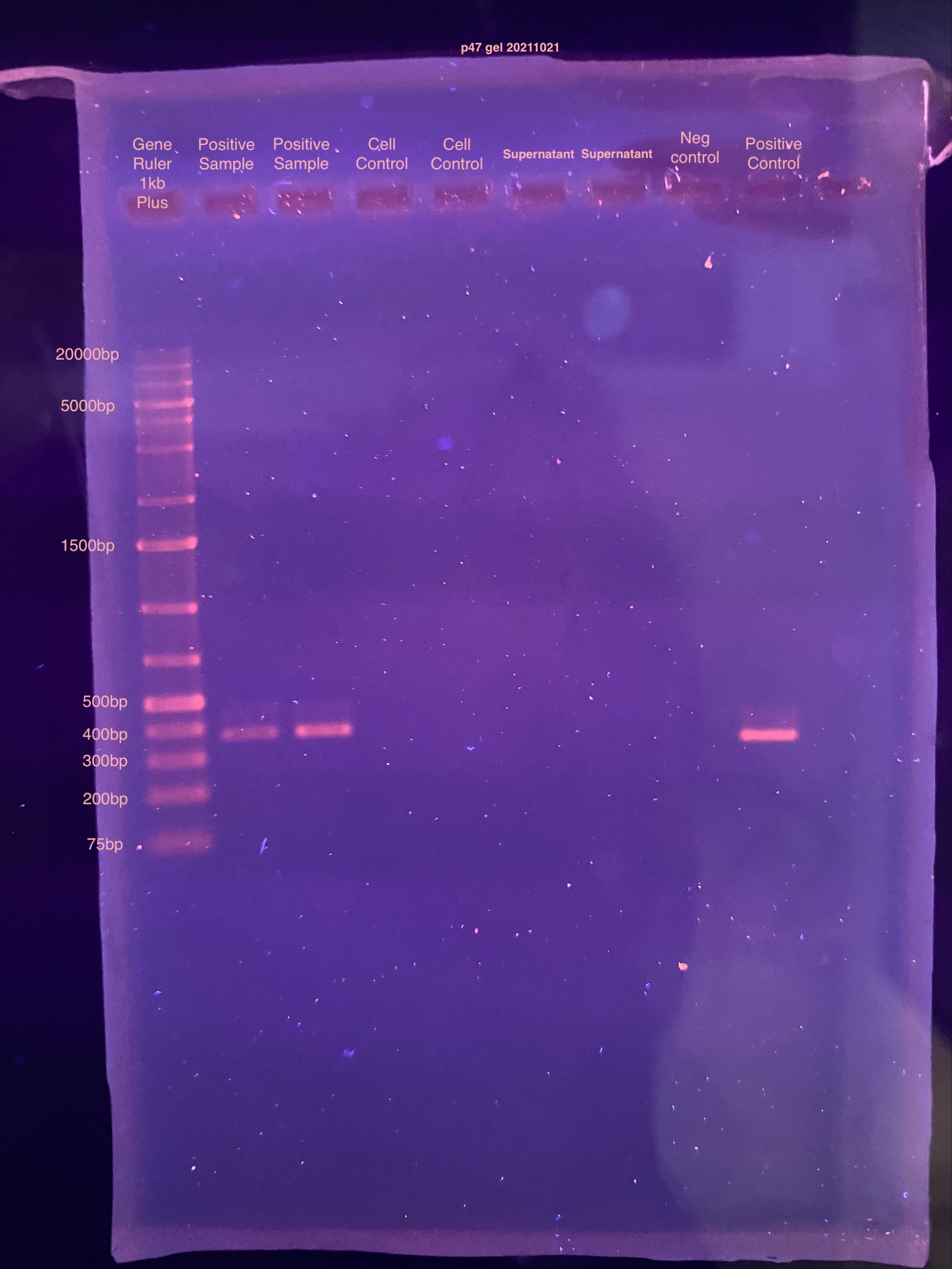
PCR worked! Only the DNA extracts from the positive samples amplified for DiNV, as well as the positive control from Kent. This could mean there really isn’t DNA in the supernatant, or that because the supernatant liquid is not compatible with the PCR reaction. I think both could be equally likely.