Oxford Nanopore minION Sequencing of pentagona DNA for Genome Building
Using the Oxford Nanopre minION for whole genome sequencing of the HMW pentagona libraries. Using the Oxford Nanopore Ligation Sequencing Kit and the Flow Cell Wash Kit for multiple loading and washing of the minION flow cell.
20200121 Loading first library and starting the experiment
- Opened minKNOW software on the sequencing computer
- Plugged in minION
- There was trouble recognizing the minION, had to re-install the software (but not update it)
- Clicked on the device when it finally showed up: checked the hardware to see if it was working
- Determined how much library to add to each sequencing run. Protocol says 5-50 fmols of DNA is ideal. Used the NEB calculator to convert from ng of DNA to fmols of DNA ends. Guesstimated that the library size was around 30,000bp, and used the ng/µl from the previous quantification of 42.4ng/µl (library PN2 is basically the same concentration). This ended up being 4.596 fmols of DNA per µl. Previous sequencing we did 20-25 fmols of DNA per loading and it worked well, so I wanted to do the same thing again. I chose 5µl which would be about 22 fmols of DNA per loading
- Thawed sequencing buffer (SQB), loading beads (LB), flush tether (FLT), elution buffer (EB), and one new tube of flush buffer (FB) at room temp then once thawed placed on ice
- Vortexed and spun down SQB, EB, and FB
- Pipette mixed FLT
- Took out fresh flow cell from fridge
- Removed test cell from the minION and plugged in the new flow cell
- Clicked on the device on the minKNOW software and clicked “check flow cell”
- took longer than 300 seconds to get to 37 degrees C
- 1288 pores available
- Slid open the priming port
- Made sure there wasn’t an air bubble in the priming port:
- Took a p1000 and set it to 200µl
- Jammed (gently) the tip into the priming port completely vertical
- Very carefully slowly turned the knob on the pipette (while in the locked position) to about 220µl
- Watched the pipette tip to see a small volume (10-20µl) of liquid come back up into the tip
- Quickly removed the tip without adding a bubble didn’t stay at the bottom of the tip like previous times but jumped up, didn’t seem to add a bubble though
- Prepared priming mix: added 30µl flush tether to the new tube of flush buffer. Pipetted to mix
- Added 800µl of the priming mix to the priming port (other port staying closed) with vertical p1000 tip very slowly, watching as carefully as possible to see if any bubbles showed up) most of the liquid at this point goes into the waste channel, but you can kinda of imagine it going through the channel, past the electrodes, over the sequencing array, and back around into the waste channels
- Waited 5 minutes
- Pipette mixed loading beads with p200 set to ~150 right before adding beads, mix again with it set to the right volume for extra thorough mixing
- Prepared library in DNA lo bind tube:
- 5µl PN1 library
- 7µl elution buffer
- 37.5µl sequencing buffer
- 25.5µl just pipette mixed loading beads
- After the 5 minutes opened the SpotON port
- Added another 200µl of priming mix with a p1000 tip to the priming port very slowly at this step you can see a small bubble of priming mix come out of the SpotOn port every time you depress the pipette, it’ll recede back down in the sequencing array
- Mixed the prepared library with a wide bore p200 pipette
- Using regular p200 pipette tip added 75µl library (all) to the SpotON port by dripping, watching to see each drop recede down over the array before adding the next one
- Closed SpotON port, the bung in the hole
- Closed the priming port
- Closed the lid of the minION
- On the minKNOW software clicked New Experiment
- Named experiment pentagonaJan2020
- Named sample PN1
- Set sequencing kit to the appropriate one, no expantion packs for barcoding
- Set to high accuracy basecalling
- Save all data to new folder called pentagonaJan2020
- Didn’t change any other settings on the interface
- Pressed start!
- Initial mux scan: 1280 pores available
- Started ~11am
11pm 20200121 Washing and Re-loading the minION
Nuclease Wash
- Thawed wash solution B at room temp (from wash kit), vortexed, spun down, then placed on ice
- Put wash solution A on ice
- When solution B was thawed, made wash mix in a DNA lo bind tube
- 380µl wash solution B
- 20µl wash solution A
- Pipetted to mix (contains DNase)
- Paused sequencing run on minKNOW software
- Opened lid and made sure priming and SpotON ports were still closed
- Removed all liquid from waste port 1 with a p1000 pipette, it was a little over 1mL of liquid. Pipetted completely vertically
- Opened priming port
- Again drew back liquid in priming port to remove bubbles:
- Took a p1000 and set it to 200µl
- Jammed (gently) the tip into the priming port completely vertical
- Very carefully slowly turned the knob on the pipette (while in the locked position) to about 220µl
- Watched the pipette tip to see a small volume (10-20µl) of liquid come back up into the tip there looked like there could be a bubble in the channel right above the priming port, but it did not move, either when doing this or when adding liquids in further steps. I left it because there wasn’t really anything else I could do
- Quickly removed the tip
- Added 400µl of the wash mix to the priming port slowly without adding a bubble at this step you can see the previous library come off the flowcell and get flushed into the waste channel because the white loading beads move through the channels
- Closed priming port
- Waited 30 minutes for the nucleases to work
- Then made sure that the priming port and the SpotON port were closed
- Removed all the waste liquid from the waste channel using waste port 1 and a p1000 pipette, again it was about 1mL
Reload Library
- Followed same steps as above for priming and loading a library
- Used same library PN1 with the same amount of library because it seemed to be sequencing well with that amount of DNA
- After closing the lid pressed resume in the minKNOW software
- Immediately did a mux scan and showed came back with 1079 active pores
1:45pm 20200123 Washing and Reloading Library a Second Time
- minION only sequencing at 20% so it was time to re-load
- Paused run and prepared washing solutions just as perviously
- When opening priming port it looked like there was a bubble again, but this time it came out when doing the pipetting suck up of liquid
- Followed same wash procedure as above
- Prepared new library to load: this time used the PN2 library. But because the quantifications were basically the same, I used the same volume of library as for PN1
- Followed same priming and loading steps steps as above
- Noticed a bubble in the channel by the electrodes when adding the second 200µl of priming mix to the priming port, but it did not move and there is no way to deal with them that far in
- Resumed sequencing run
5:45am 20200124 Washing and Reloading Library a Third Time
- Again poor sequencing and many unavailable pores so it was time to wash and reload
- Followed steps as above to wash out the flow cell
- Followed same steps as above to prepare a new library with the PN2 library
Issues with reloading this time:
- Adding 800µl priming mix went fine, the problem occurred when the SpotON port was opened
- When adding 200µl of priming mix to the priming port when the SpotON port was open, it did not seem to go down into the sequencing array: I immediately noticed that the small bubble of liquid that usually pushes out the SpotON port when adding in this step would not recede back down into the array, it would just stay as a bubble on the top
- I stopped adding about 20µl in, and put the rest of the priming mix back into the tube
- At this point I didn’t know exactly what to do. there are recommendations on the Oxford Nanopore community forum for what to do if your library won’t go down the SpotON port, which seemed like a similar issue so I at first tried to follow those troubleshooting recommendations:
- I removed all the waste from the waste channel via waste port 1 with both ports closed, then I flushed it with 1mL of nuclease free water and removed that as well. This did nothing
- I added about 20µl of priming mix to the top of the SpotON port, it stayed there, then I stuck a p1000 set at 200µl in the priming port and drew back from there and watched the bubble go down from the SpotON port. I tried this twice and it still didn’t release whatever blockage
- Finally with recommendations from Jon I added the 200µl of priming mix to the priming port regardless of the bubble. A large bubble formed over the SpotON port but I sucked up whatever was there hoping that some went down. I then mixed then added the library to the SpotON port, as expected it did not go down. I then drew back from the priming port to get it to go into the flow cell. I could see the loading beads go back up the channel towards the electrodes, but it also looked like some of it got over the sequencing array.
- I closed it and resumed the run at this point and it came back with 886 pores!
9pm 20200124 Washing and Reloading Library a Fourth Time
- We had more library, so we decided to try to squeeze as much as we possibly could out of this flow cell and try adding again
- Again poor sequencing and a majority unavailable pores
- Followed steps as above to wash out the flow cell
- This time no blockage! There seemed to be a bubble that zoomed out towards the waste channel when adding the 800µl priming mix, so that must have cleared the blocking
- This time prepared a library with what was left of the libs we had: 3.5µl of each (adding up to 7) then 5µl of elution buffer. The rest of the prep followed the same as above
- It came back sequencing with more than 600 pores
- This was run until the 72 hours of sequencing was up: we got 2.5 million reads!
Final charts in the minKNOW software :
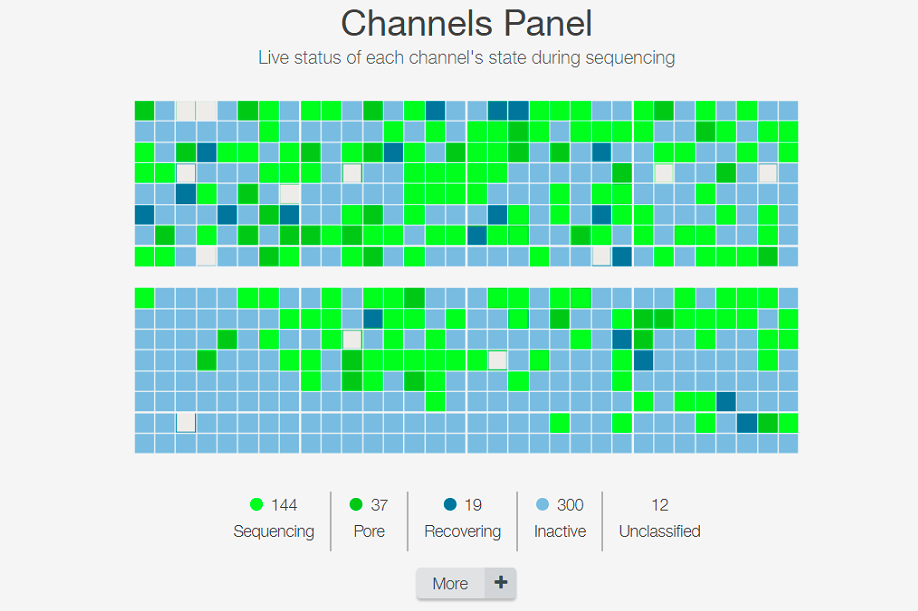
Channels being sequenced: very few sequencing at the end but still has some output!
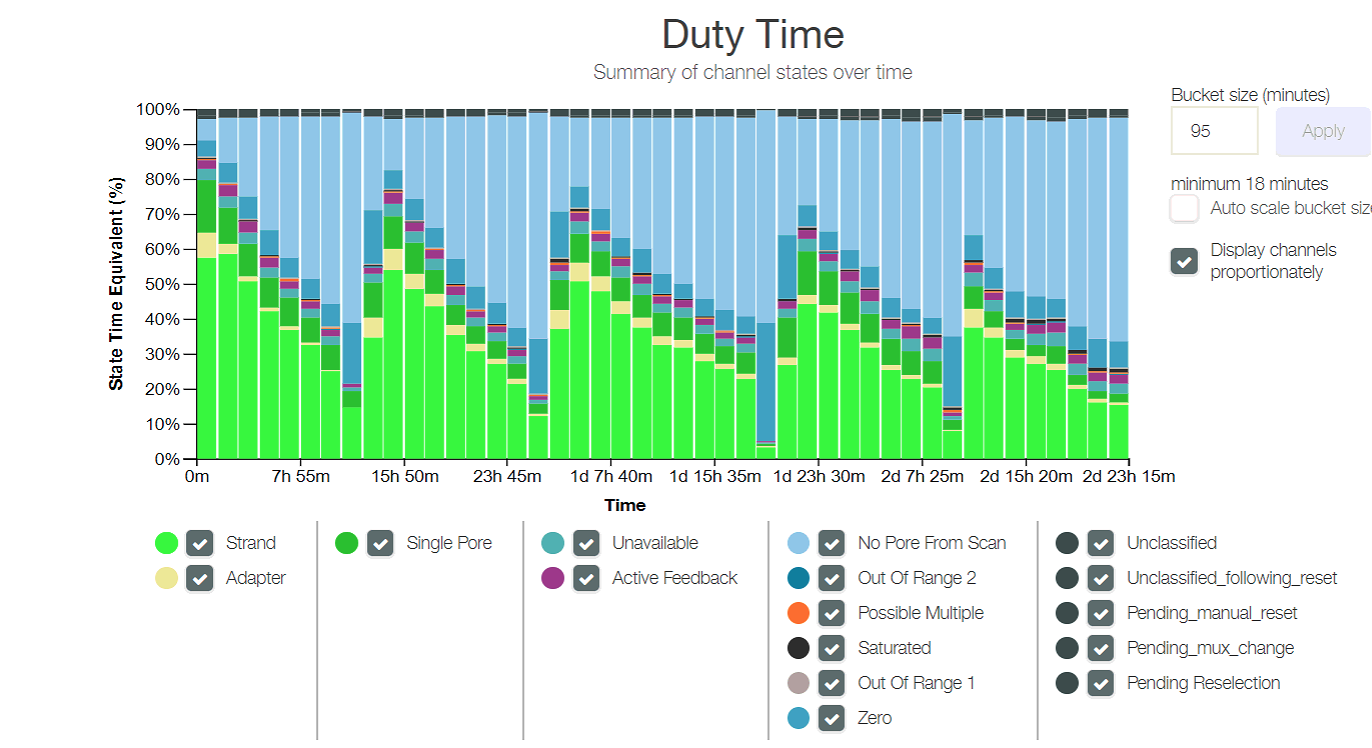
Duty time plot of the entire sequencing run, showing percentages of how many pores were in what mode across buckets of time. It is relatively consistent across additions of libraries and washes, and shows each time the sequencing slowed and then ramped up again after a wash and reload
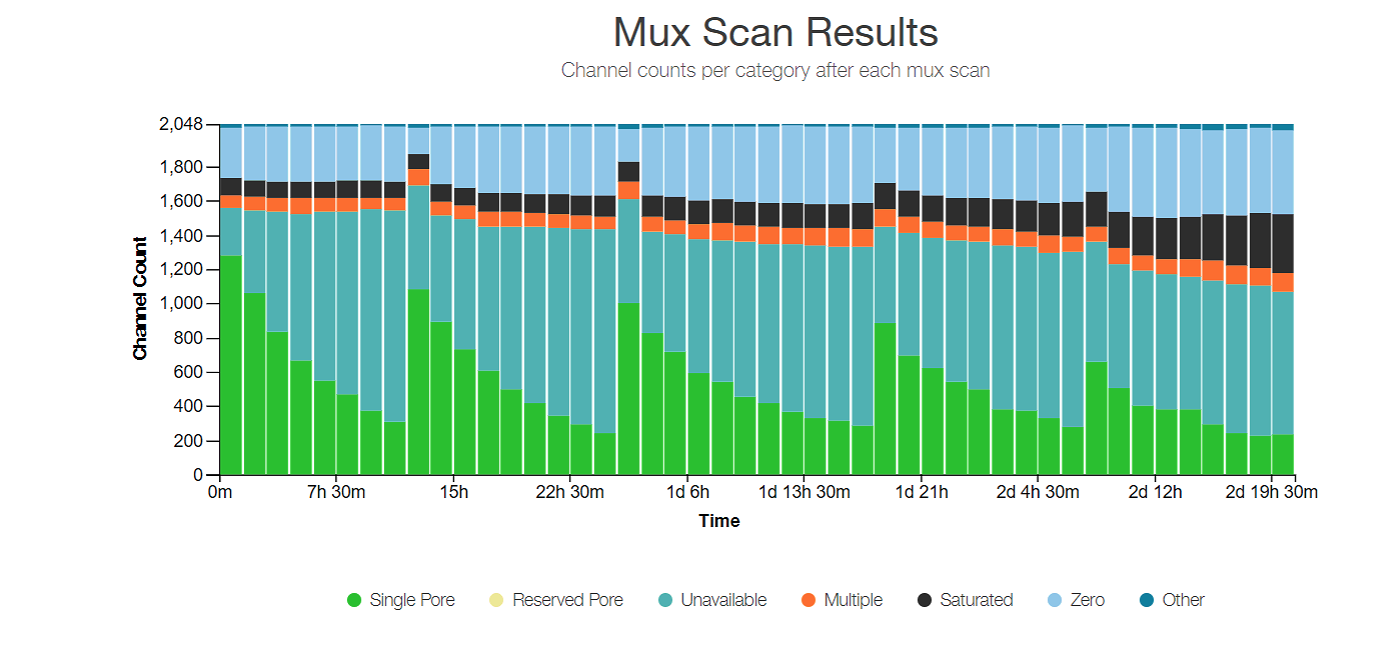
Similar plot, shows the result of the mux scan (multiplex scan) that determines how many pores to use to sequence at one time. Only about a fourth of the pores are used at once, and there are 4 pores around one electrical channel
Histogram of read counts over the whole sequencing run. Obviously skewed to smaller reads
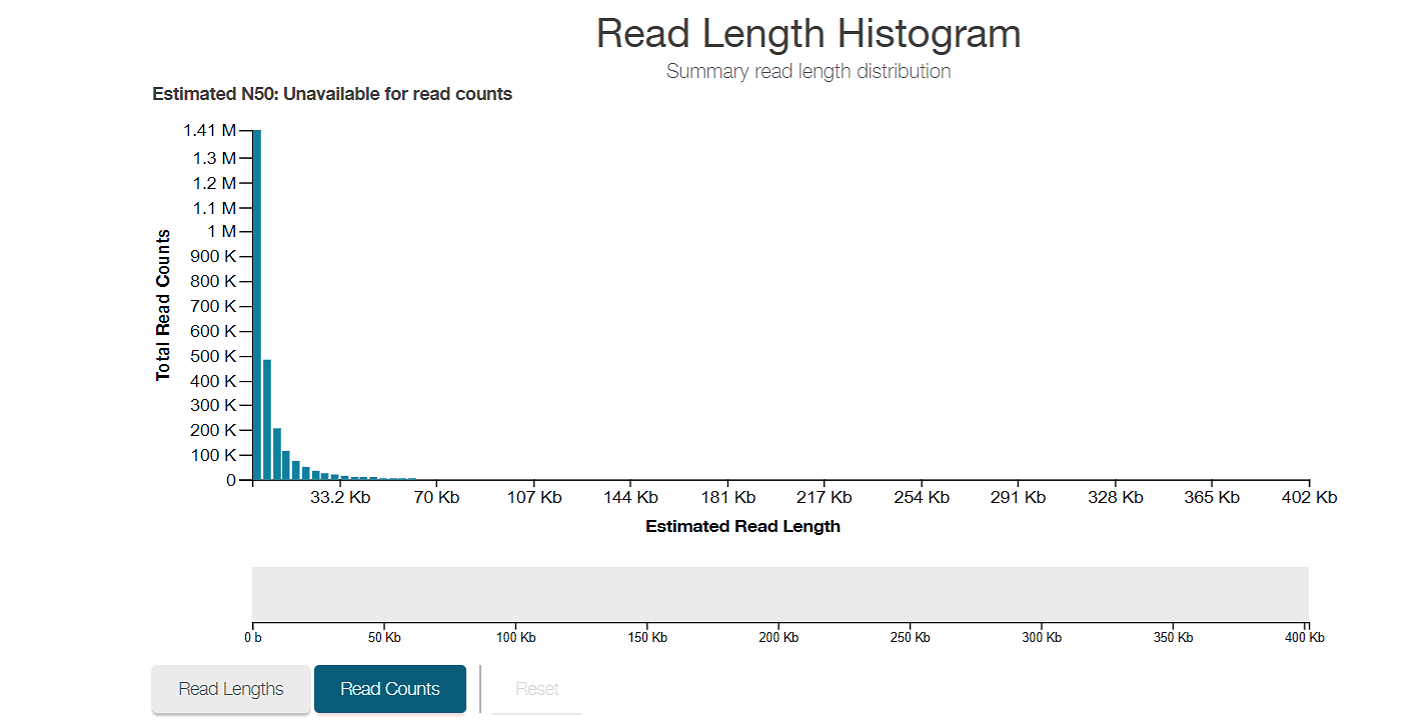
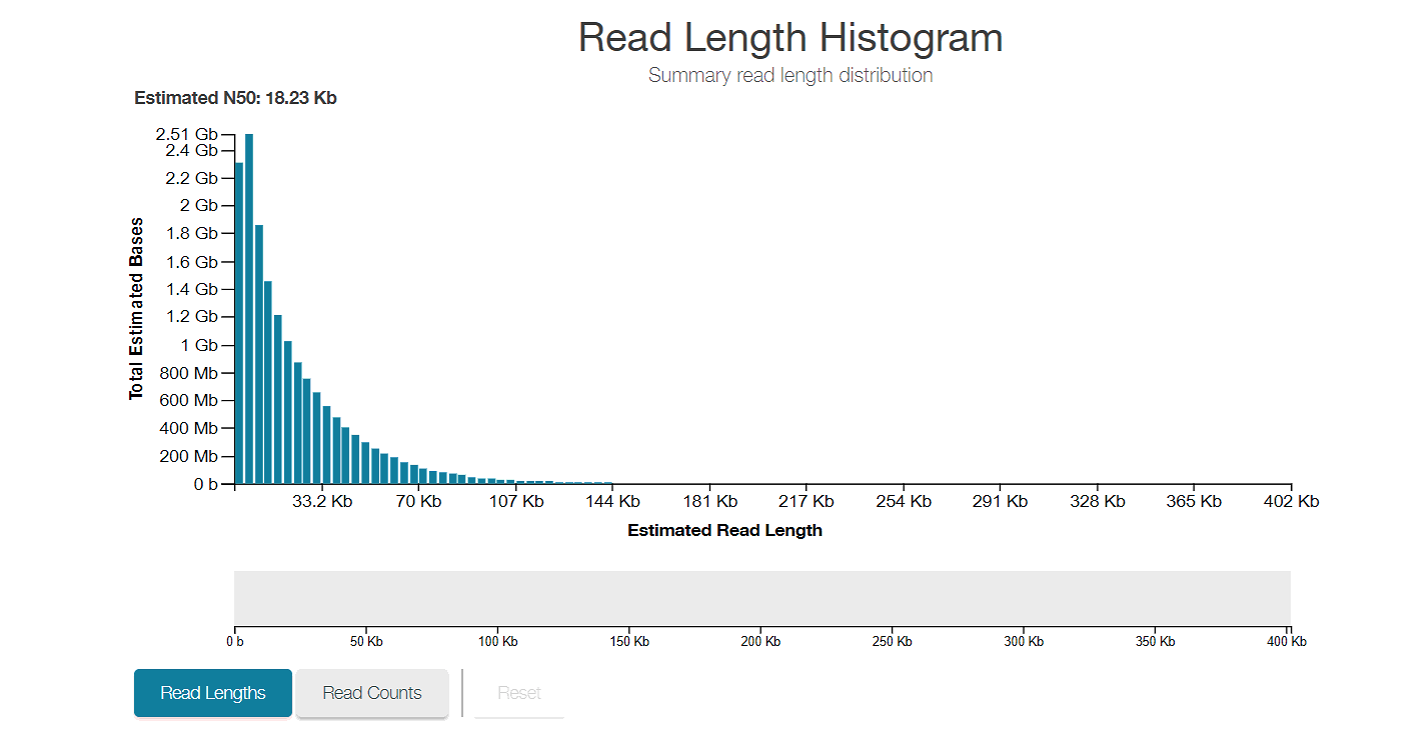
Histogram of read lengths over the whole sequencing run, using total estimated bases instead of counts as the y axis. This allows for the fact that longer reads contain more information than smaller reads to be taken into account in your estimation of how well the sequencing is going. 100 reads of 5kb length is less information than 20 reads of 30kb length. We got an N50 of 18.23kb!
{kind=link}
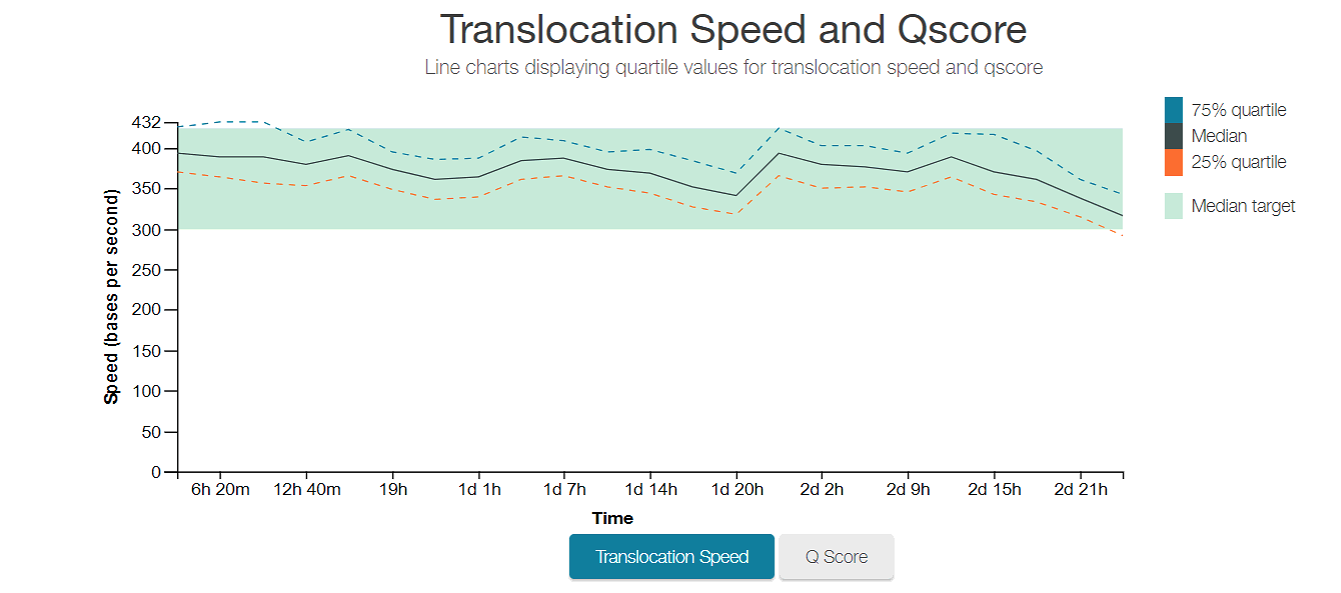
Plot of the translocation speed, or number of bases that go through the pores in 1 second, over the whole sequencing run. You want it to stay in that green range
{kind=link}
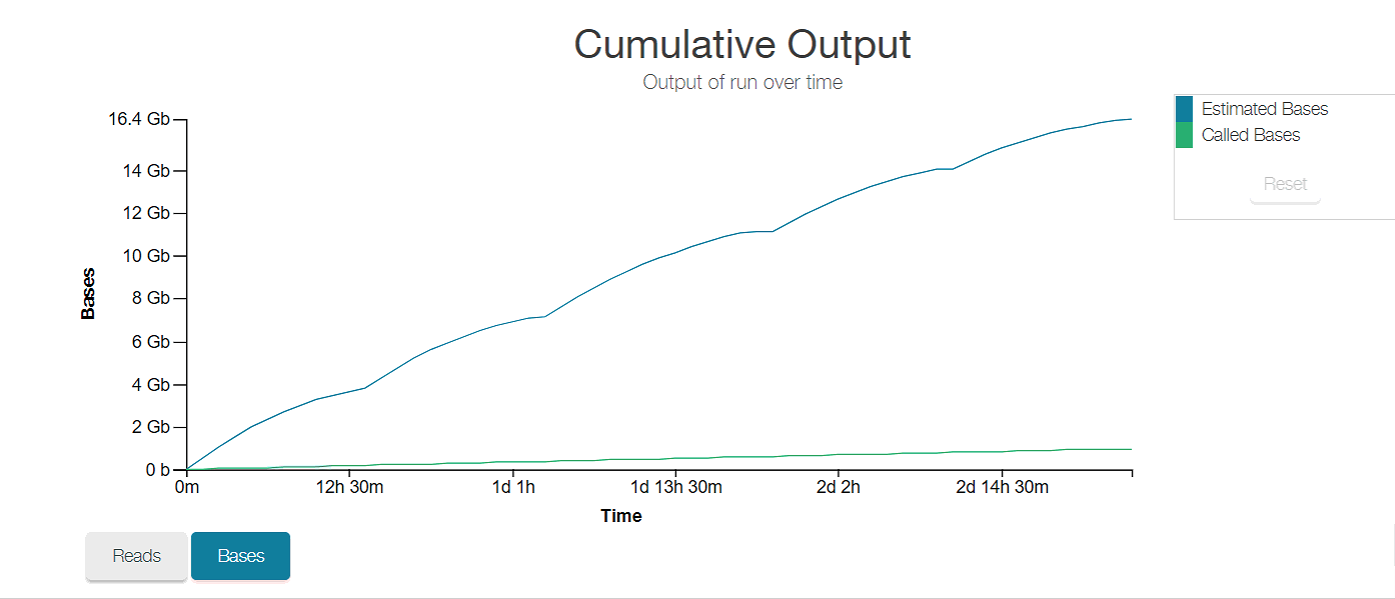
Final cumulative output plot: WOW! You can clearly see when sequencing effort plateaus, and how it picks back up again exponentially with each wash. And so many reads!
{kind=link}