Block 10 Stranded mRNA Library Prep
Stranded mRNA Library Prep for CASE EecSeq Block 10
Following half reactions from the KAPA Biosystems Stranded mRNA-Seq Kit, also see the EeqSeq protocol
Capture and Fragmentation
- Calculated number of mRNA capture beads needed for 12 samples
- 26.25*12= 315μl beads
- Pipetted 315μl mRNA capture beads into a new 1.5mL tube and put on the large magnet stand
- Removed supernatant
- Added 315μl of bead binding buffer and pipetted to mix off magnet
- Put back on magnet and removed as much supernatant as possible
- Added 315μl of bead binding buffer again and pipetted to mix off magnet
- Put back on magnet and again removed as much supernatant as possible
- Added 315μl of bead binding buffer and pipetted to mix off magnet
- Prepared RNA samples: 1μg in 25μl with the rest made up with nuclease free water
| Sample | RNA qubit | volume for 1000ng | water to 25ul |
|---|---|---|---|
| B10T24J1 | 71 | 14.1 | 10.9 |
| B10T24J2 | 50 | 20.0 | 5.0 |
| B10T24J3 | 81.5 | 12.3 | 12.7 |
| B10T24J4 | 52 | 19.2 | 5.8 |
| B10T24J5 | 100 | 10.0 | 15.0 |
| B10T24J6 | 80.5 | 12.4 | 12.6 |
| B10T24J7 | 42.2 | 23.7 | 1.3 |
| B10T24J8 | 49.5 | 20.2 | 4.8 |
| B10T24J9 | 99.8 | 10.0 | 15.0 |
| B10T24J10 | 88.5 | 11.3 | 13.7 |
| B10T24J11 | 98 | 10.2 | 14.8 |
| B10T24J12 | 48.8 | 20.5 | 4.5 |
- Added 25μl of the resuspended mRNA capture beads to each sample tube
- Placed tubes in the 1st mRNA capture program in the thermocycler
- Placed tubes on the magnet plate and removed all supernatant when the solution went clear
- Removed tubes from the magnet plate and resuspended beads in 100μl of bead wash buffer
- Placed tubes on the magnet plate and removed all of the supernatant when the solution went clear
- Resuspended beads in 25μl of RNase-free water off magnet
- Placed tubes in the 2nd mRNA capture program in the thermocycler
- Took tubes out of the thermocycler and added 25μl of bead binding buffer to each tube and pipetted to mix
- Incubated the tubes are 20C for 5 minutes (program in thermocycler for this)
- Made 1X Fragment, Prime, and Elute Buffer on ice bucket:
- 5.5μl nuclease free water * 12.5 = 67.1μl
- 5.5μl 2X FPE buffer * 12.5 = 67.1μl
- Placed tubes on the magnet plate and removed supernatant when the solution went clear
- Resuspended beads off magnet in 11μl 1X FPE buffer
- Put in thermocycler for RNA fragmentation program (7 minutes ate 94C)
- Note: at this step, a master mix for the 1st strand synthesis was supposed to have been made, however I used 2nd strand reagents on accident. 5μl of 2nd strand reagents were added to samples J01-J08. These samples were frozen at -80 while samples J09-J12 were carried through 1st and second strand synthesis. See J01-J08 progress below
First and Second Strand Synthesis - Made correct 1st strand synthesis master mix for 4 samples
- 5.5μl 1st strand synthesis buffer * 4.2 = 23.1μl
- .5μl KAPA script * 4.2 = 2.1μl
- IMMEDIATELY placed tubes on magnet plate once the program was finished
- Removed 10μl of clear supernatant and placed in new PCR strip tubes on ice
- Added 5μl of the 1st strand synthesis master mix to each tube and pipetted to mix
- Placed in thermocycler 1st strand synthesis program
- Made 2nd Strand Synthesis and Marking Master Mix on ice:
- 15.5μl 2nd strand marking buffer * 4.2 = 65.1μl
- 1μl second strand enzyme * 4.2 = 4.2μl
- Removed tubes from the thermocycler and placed on ice
- Added 15μl of the 2nd strand synthesis and marking master mix and pipetted to mix
- Put in thermocycler 2nd strand synthesis program
- Took KAPA Pure Beads out of the 4 degree
- Took tubes out of the thermocycler and added 54μl KAPA Pure Beads, pipetting to mix
- Incubated tubes on shaker for 15 minutes at room temp
- Made A-tailing Safe Stopping Point Master Mix on ice:
- 6.75μl water * 4.2 = 28.35μl
- 0.75μl 10X A-tailing buffer * 4.2 = 3.15μl
- Performed normal bead clean up with fresh 80% EtOH
- Resuspended the beads in 7.5μl of the A-tailing Safe Stopping Point Master Mix
- Spun tubes down to make sure all the beads were off the sides
- Placed tubes in 4 degree overnight
Salvaging Samples J01-J08
Recommendations from KAPA technical service: 3X cleanup, elute in 1X FPE buffer, 1 minute incubation at 65C, then proceed with 1st strand synthesis
- 3X cleanup, 15μl in each tube, so 45μl KAPA pure beads added to each sample
- Performed normal bead cleanup extra carefully
- Made 1X FPE buffer:
- 5.5μl nuclease free water * 8.5 = 67.1μl
- 5.5μl 2X FPE buffer * 8.5 = 67.1μl
- Elute and Resuspend beads in 11μl 1X FPE buffer and place on magnet
- Save 10μl of supernatant
- Placed tubes in the thermocycler at 65C for 1 minute
- Placed tubes on ice
- Made 1st strand synthesis master mix
- 5.5μl 1st strand synthesis buffer * 8.5 = 46.75μl
- .5μl KAPA script * 8.5 = 2.75μl
- Added 5μl of the 1st strand synthesis master mix to each tube and pipetted to mix
- Placed in thermocycler 1st strand synthesis program
- Made 2nd Strand Synthesis and Marking Master Mix on ice:
- 15.5μl 2nd strand marking buffer * 8.5 = 131.75μl
- 1μl second strand enzyme * 8.5 = 8.5μl
- Removed tubes from the thermocycler and placed on ice
- Added 15μl of the 2nd strand synthesis and marking master mix and pipetted to mix
- Put in thermocycler 2nd strand synthesis program
- Took KAPA Pure Beads out of the 4 degree
- Took tubes out of the thermocycler and added 54μl KAPA Pure Beads, pipetting to mix
- Incubated tubes on shaker for 15 minutes at room temp
- Made A-tailing Safe Stopping Point Master Mix on ice:
- 6.75μl water * 8.5 = 56.03μl
- 0.75μl 10X A-tailing buffer * 8.5 = 6.225μl
- Performed normal bead clean up with fresh 80% EtOH
- Resuspended the beads in 7.5μl of the A-tailing Safe Stopping Point Master Mix
- Placed in 4 degree until later that day
A-tailing and Adapter Ligation
All 12 samples now going forward
- Made A-tailing after safe stopping point master mix
- 5.25μl nuclease-free water * 12.5 = 65.625μl
- 0.75μl A-tailing buffer * 12.5 = 9.375μl
- 1.5μl A-tailing enzyme * 12.5 = 18.75μl
- Took sample tubes out of the 4 degree and added 7.5μl of the A-tailing after safe stopping point master mix to each tube and pipetted to mix
- Put tubes in the thermocycler A-tailing program
- Made the Adapter Ligation Master Mix:
- 8μl nuclease-free water * 12.5 = 100μl
- 7μl ligation buffer * 12.5 = 87.5μl
- 2.5μl DNA ligase * 12.5 = 31.25μl
- Added Ligation master mix and planned adapter to each sample, pipetted to mix:
| Sample | μl of LMM | μl of Adapter (700μM) |
|---|---|---|
| B10T24J1 | 17.5 | 2.5 SAII_NO_N |
| B10T24J2 | 17.5 | 2.5 SAII_NO_N |
| B10T24J3 | 17.5 | 2.5 SAII_NO_N |
| B10T24J4 | 17.5 | 2.5 SAIIv2 |
| B10T24J5 | 17.5 | 2.5 SAII_NO_N |
| B10T24J6 | 17.5 | 2.5 SAIIv2 |
| B10T24J7 | 17.5 | 2.5 SAIIv2 |
| B10T24J8 | 17.5 | 2.5 SAIIv2 |
| B10T24J9 | 17.5 | 2.5 SAIIv2 |
| B10T24J10 | 17.5 | 2.5 SAIIv2 |
| B10T24J11 | 17.5 | 2.5 SAII_NO_N |
| B10T24J12 | 17.5 | 2.5 SAII_NO_N |
- Placed tubes on shaker for 30 minutes at room temp
- Added 35μl of room temperature PEG to each sample and pipetted to mix
- Performed normal bead clean up with fresh 80% EtOH
- Resuspended beads in 25μl 10mM Tris HCl pH8
- Added 25μl of room temperature PEG to each tube and pipetted to mix
- Performed normal bead cleanup with fresh 80% EtOH
- Resuspended and eluted beads in 11μl 10mM Tris HCl pH8 and placed on magnet plate
- Removed 10μl of supernatant into new PCR tubes and placed in the freezer at -20 for the weekend
Library Amplification and Index Addition
- Set up amplification with KAPA Hot start ready mix and individual index pairs, added to the 10μl of sample. Reactions set up on ice
| Sample | μl of HSRM | μl of Index 1 | μl of Index 2 |
|---|---|---|---|
| B10T24J1 | 12.5 | 1.25 501 | 1.25 701 |
| B10T24J2 | 12.5 | 1.25 502 | 1.25 702 |
| B10T24J3 | 12.5 | 1.25 503 | 1.25 703 |
| B10T24J4 | 12.5 | 1.25 501 | 1.25 701 |
| B10T24J5 | 12.5 | 1.25 504 | 1.25 704 |
| B10T24J6 | 12.5 | 1.25 502 | 1.25 702 |
| B10T24J7 | 12.5 | 1.25 503 | 1.25 703 |
| B10T24J8 | 12.5 | 1.25 504 | 1.25 704 |
| B10T24J9 | 12.5 | 1.25 505 | 1.25 705 |
| B10T24J10 | 12.5 | 1.25 506 | 1.25 706 |
| B10T24J11 | 12.5 | 1.25 505 | 1.25 705 |
| B10T24J12 | 12.5 | 1.25 506 | 1.25 706 |
- Vortexed and spun down
- Samples J01-J08 were placed in the thermocycler PCR program for 14 cycles to compensate for inevitable loss, samples J09-J12 were placed in the thermocycler with the usual 12 cycle program
- After the PCR, 25μl of KAPA Pure beads (1X) was added to each sample and pipetted to mix
- Performed normal bead clean up with fresh 80% EtOH
- Resuspended and eluted beads in 22μl 10mM Tris HCl pH 8
QC
High Sensitivity Qubit
| Sample | Standard 1 | Standard 2 | Average DNA(ng/μl) |
|---|---|---|---|
| B10T24J1 | 46 | 26006 | 32.2 |
| B10T24J2 | - | - | 31.8 |
| B10T24J3 | - | - | 46.2 |
| B10T24J4 | - | - | 33.9 |
| B10T24J5 | - | - | 22.6 |
| B10T24J6 | - | - | 31.4 |
| B10T24J7 | - | - | 29.2 |
| B10T24J8 | - | - | 22.8 |
| B10T24J9 | - | - | 80.4 |
| B10T24J10 | - | - | 86.8 |
| B10T24J11 | - | - | 62.8 |
| B10T24J12 | - | - | 60.4 |
D5000 TapeStation
See full report here
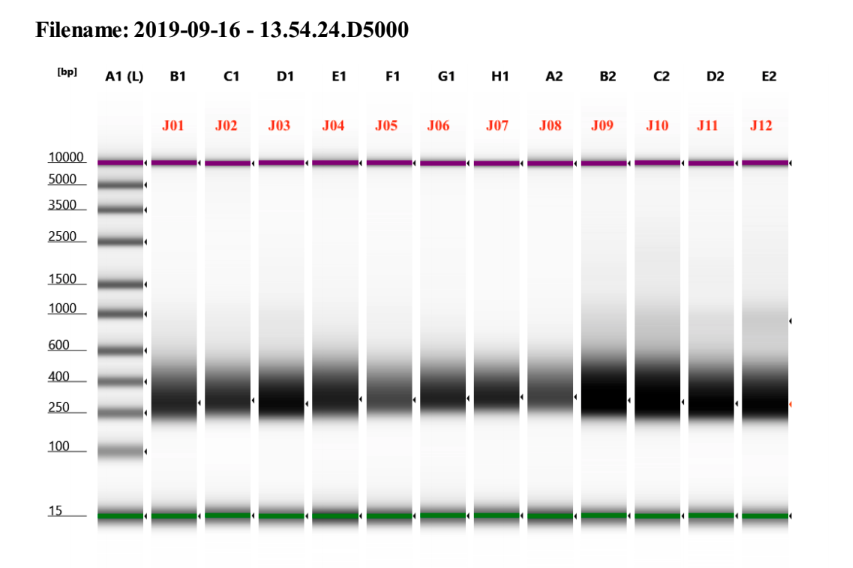
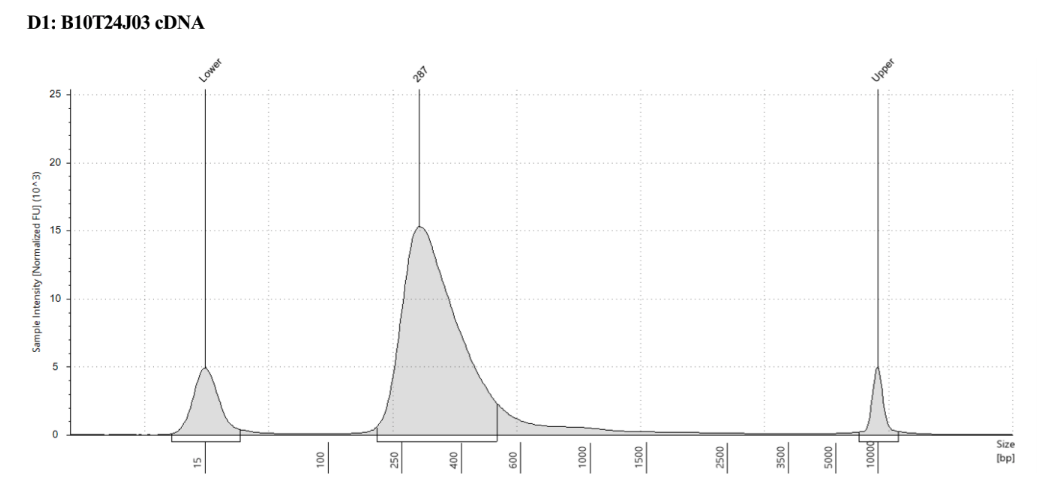
Representative sample trace:
Written on September 16, 2019